Antibody–Antigen Docking
Antibodies are essential recognition elements of the immune system, which bind tightly and specifically to various antigens to block their activity or to mark them for destruction. Antibodies are increasingly used as diagnostic tools and therapeutic drugs, and an important component of the antibody design/engineering process is the availability of high quality three-dimensional structure of antibody–antigen complexes. X-ray crystallography provides the most accurate and detailed data on protein structure and interactions, however co-crystallization of antibody–antigen complexes remains a major challenge, and the number of antibody structures determined in complex with their antigens is still relatively small.
By contrast, computational modeling provides a fast and inexpensive route for generating structural models for such complexes. The variable domains (Fvs) of the heavy and light chain are of special interest in computational modeling, as they typically impart most or all of the specificity of an antibody for its antigen target. Our modeling protocol takes into consideration the most updated scientific knowledge of antibody–antigen interactions as well as conformational flexibility of the CDR region (especially the H3 loop), and utilizes state-of-the-art docking software tools to find the relative transformation and conformation of the antibody and antigen involved in energetically favorable complex formation. The general procedures are: First, a rigid body global search is performed and geometrically plausible antibody–antigen complex models are generated. In this process, user-defined restraints (obtained from experimental data) can be applied to limit the search space. The resulting models are scored and ranked using a scoring function, developed specifically for antibody systems. The best-scored structures are then clustered and representatives of the largest clusters are selected for structural optimization by energy minimization before being presented to the customer.
Antibody–antigen docking process
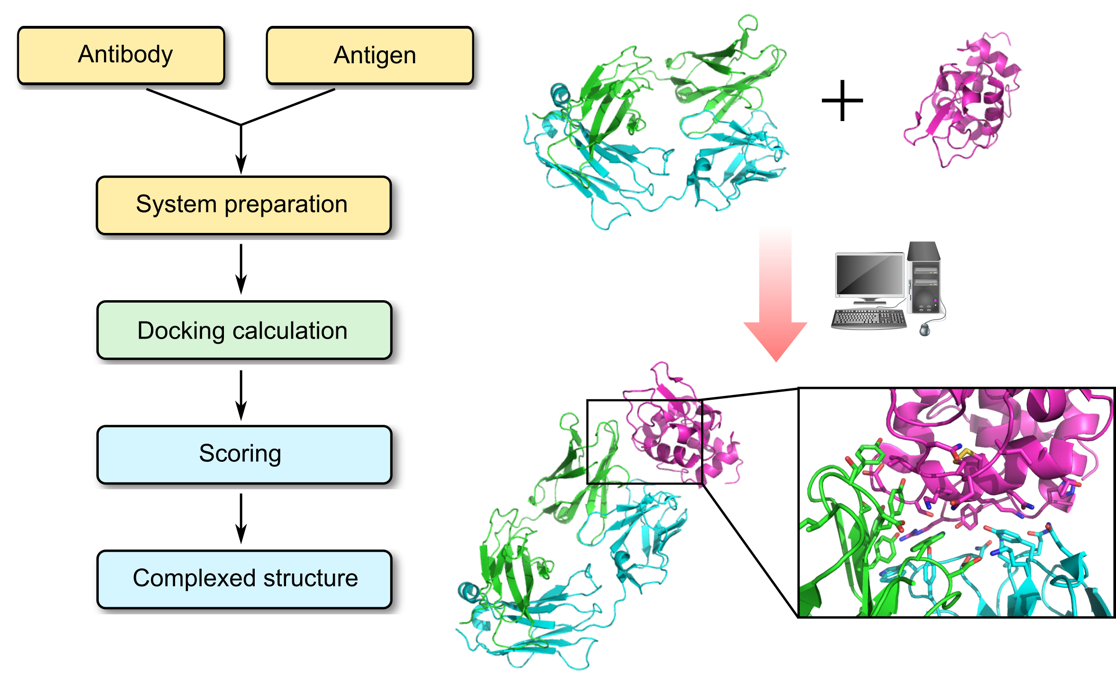
Profacgen employs computational docking techniques to search all possible binding modes in the translational and rotational space between an antibody (Fv, Fab, or full antibody) and its antigen, with modeling of conformational changes of the variable loops upon complex formation. This is an important means for understanding the physicochemical forces that underlie macromolecular interactions, and a working hypothesis can be build based on the docked complex, guiding further experiments for the rational design of antibody-based therapeutics.
Features:
Scoring function dedicated to assessing antigen–antibody interactionsSolvated docking protocol allowing explicit inclusion of interfacial water molecules
Filtering output predictions based on user-defined restraints
Clustering to find highly populated clusters of low-energy conformations
Support modified amino acids in antibody and antigen
Modeling and refinement of Fv loop regions
Computational epitope mapping
Compatible with common antibody numbering schemes: Chothia, Kabat, IMGT, Aho
Model quality assessment by multiple criteria
We provide the service in a customizable fashion to suit our customers’ specific research goals. Please do not hesitate to contact us for more details about our antibody–antigen docking service.
Fill out this form and one of our experts will respond to you within one business day.