The discovery of new pharmaceutical drugs remains one of the most demanding tasks in biomedical research. Traditional high-throughput screening (HTS) relies on blind testing of molecules sourced from nature or synthesized in laboratories, often resulting in high costs and low hit rates. As a powerful alternative, virtual screening (VS) identifies hit molecules from large compound collections in a fraction of the time, reducing the number of compounds that must be synthesized, purchased, or tested while expanding chemical space through virtual libraries. VS has become a crucial step in early-stage drug discovery, and ultra-large virtual screening—powered by cloud computing and enormous compound collections—is rapidly emerging as the new standard.
Profacgen's Virtual Screening platform leverages state-of-the-art VS technologies and software tools to drive hit and lead identification. Our optimized protocols reduce experimental library size, increase the probability of discovering novel hits faster and more economically, and mitigate the risk of lead optimization failure caused by ADMET deficiencies—delivering a comprehensive, customizable solution for structure-based and ligand-based drug design.
Virtual screening refers to the computational assessment of large compound libraries to identify small molecules most likely to bind to a drug target, typically a protein receptor or enzyme. Unlike physical high-throughput screening that requires synthesis or procurement of compounds and robotic automation, virtual screening operates entirely in silico, evaluating chemical structures through molecular descriptors, force-field calculations, and statistical models. The methodology traces its conceptual origins to Hansch analysis in the 1960s but gained practical momentum in the late 1990s with the advent of affordable computational resources, growing structural databases, and refined scoring algorithms.
Two principal paradigms dominate virtual screening: ligand-based virtual screening (LBVS), which exploits known active compounds to discover chemically similar or pharmacologically equivalent molecules, and structure-based virtual screening (SBVS), which utilizes three-dimensional protein structures to evaluate complementarity between candidate ligands and binding sites. The choice between these approaches—or their strategic combination—depends on the availability of structural data, known ligands, and the specific characteristics of the biological target under investigation.
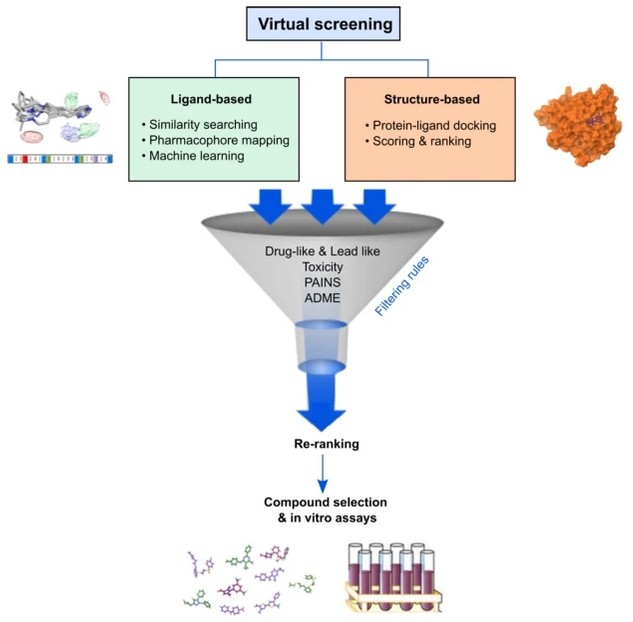
Ligand-Based Virtual Screening (LBVS)
Our LBVS platform leverages known bioactive compounds to identify structurally novel molecules with similar pharmacological properties. Services include 2D fingerprint similarity searching, 3D shape and electrostatic matching, pharmacophore modeling, quantitative structure-activity relationship (QSAR) modeling, and machine learning-based activity prediction.
Structure-Based Virtual Screening (SBVS)
Profacgen's SBVS services utilize high-resolution protein structures to evaluate binding complementarity through advanced docking algorithms, molecular dynamics simulations, and binding free energy calculations. We support apo structures, holo complexes, and homology models across diverse target classes.
Our molecular docking services provide detailed analyses of protein-ligand interaction geometries, binding pose predictions, and affinity estimations using validated scoring functions. We offer both rigid-receptor and induced-fit docking protocols with explicit water handling and cofactor inclusion.
Our hybrid approaches integrate ligand-based and structure-based methods with machine learning consensus scoring to maximize hit identification rates. Custom protocols combine pharmacophore filtering, docking refinement, and statistical ranking for challenging targets with limited structure or ligand information.
Profacgen provides comprehensive absorption, distribution, metabolism, excretion, and toxicity (ADMET) property predictions using validated quantitative models and machine learning classifiers. Early identification of pharmacokinetic liabilities and safety concerns streamlines lead optimization prioritization.
Quantum Mechanics (QM)-Based Drug Design
For targets where classical force fields prove insufficient, our QM-based approaches employ ab initio and DFT calculations to accurately model ligand binding energetics, reaction mechanisms, and electronic polarization effects in enzyme active sites, delivering superior accuracy for metalloproteins and covalent inhibitors.
Background:
A biotechnology client sought chemically distinct BTK inhibitors for autoimmune disease indications, requiring selectivity over the TEC kinase family and other kinome members.
Our Solution:
Profacgen executed a hybrid virtual screening campaign combining a 3D pharmacophore model derived from crystallographic BTK inhibitor complexes with ensemble docking against 12 representative kinase DFG conformations. We screened 4.2 million compounds from the Enamine REAL database, applying a hierarchical filtering cascade of pharmacophore matching, Glide SP docking, MM-GBSA rescoring, and machine learning-based kinome selectivity prediction.
Final Results:
Among 72 selected compounds for purchase and biochemical testing, 18 exhibited BTK IC50 values below 100 nM (25% hit rate), with the most potent compound achieving 3.2 nM. Importantly, lead compound PFC-BTK-7 demonstrated >50-fold selectivity against TEC, ITK, and EGFR kinases, meeting the client's selectivity criteria. Structure-based optimization guided by our team improved metabolic stability from human microsomal t1/2 of 12 minutes to 87 minutes while maintaining sub-10 nM potency.
Background:
K-Ras mutations occur in approximately 25% of human cancers, yet the protein's smooth surface and picomolar affinity for GTP/GDP long rendered it "undruggable." A pharmaceutical partner engaged Profacgen to identify fragment-sized scaffolds binding the switch-II pocket (S-IIP) discovered in G12C mutant structures.
Our Solution:
We employed a structure-based virtual screening workflow specifically designed for fragment discovery: FTMap hotspot mapping to identify favorable interaction regions, pharmacophore-constrained docking of a 45,000-fragment library (MW 150-300 Da), and machine learning-based potency prediction using a support vector regression model trained on 147 known S-IIP binders.
Final Results:
Of 120 fragment hits tested by surface plasmon resonance, 38 showed measurable binding (KD < 1 mM), with 12 fragments exhibiting KD values below 100 μM. Fragment PFC-KRAS-F03 (2-chloro-4-(trifluoromethyl)benzamide) bound with 22 μM affinity and served as the foundation for a fragment-growing campaign that yielded lead compounds with single-digit nanomolar potency and cellular activity in K-Ras G12C-mutant lung cancer cell lines.
Discuss Your Virtual Screening Project
Fill out this form and one of our experts will respond to you within one business day.