We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our
Privacy Policy
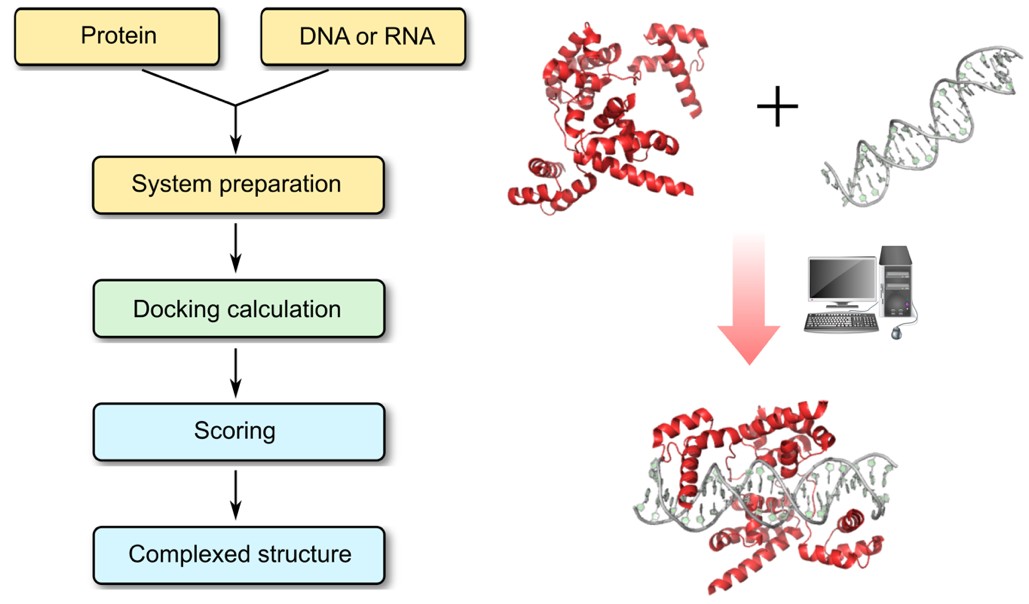
Profacgen's Protein–Nucleic Acid Docking service offers advanced computational modeling of protein interactions with DNA, RNA, and hybrid molecules. These interactions are central to key biological processes—including replication, transcription, splicing, degradation, and translation—and their dysregulation is linked to diseases ranging from neurological disorders to cancer, underscoring their importance in both basic research and therapy development.
High-resolution experimental determination of these complexes remains challenging, especially for transient or flexible systems. Computational docking provides a powerful complement, delivering atomic-level insights into recognition mechanisms. Our platform generates detailed structural models to support binding affinity prediction and the rational design of biologics targeting these critical interfaces.
Why Protein–Nucleic Acid Docking?
Understanding the structural and energetic basis of protein–nucleic acid recognition is fundamental to modern molecular biology and drug discovery. Computational docking offers several unique advantages over purely experimental approaches:
Rapid generation of structural hypotheses for protein–DNA and protein–RNA complexes without the need for crystallization or extensive sample preparation
Systematic exploration of all possible binding modes between proteins and nucleic acids, including those that undergo very large conformational changes upon binding
Integration of experimental restraints—such as mutagenesis data, cross-linking data, and footprinting results—to guide and validate docking predictions
Ability to model modified amino acids and chemically modified DNA/RNA molecules, including epigenetic modifications and therapeutic oligonucleotides
Quantitative scoring and ranking of predicted complexes using statistical potentials specifically parameterized for protein–nucleic acid interactions
Support for hybrid DNA–RNA duplexes, such as dsRNA/DNA hybrids encountered in gene editing systems and during transcription
By combining computational docking with available experimental constraints, Profacgen delivers high-confidence structural models that accelerate hypothesis-driven research and guide the rational design of engineered proteins with altered nucleic acid binding specificity.
Our Protein–Nucleic Acid Docking Service Offerings
Services
Details
Target Preparation
Protein structure curation, including homology modeling when experimental structures are unavailable
Nucleic acid model building for DNA, RNA, and hybrid DNA/RNA molecules
Support for chemically modified nucleotides and non-standard amino acids
Structural quality assessment and preprocessing for docking simulations
Binding Site Prediction & Restraint Definition
Sequence- and structure-based binding site prediction for both protein and nucleic acid partners
Integration of experimental data including mutagenesis, cross-linking, and footprinting results
Definition of blocking residues to exclude non-interacting surfaces and reduce search space
Specification of pairwise distance restraints to guide docking toward biologically relevant solutions
Protein–Nucleic Acid Docking
Rigid-body global search systematically sampling all possible binding modes
Generation of geometrically plausible complex structures filtered by user-defined restraints
Scoring using nucleic acid-specific statistical potentials (protein–RNA and protein–DNA parameter sets)
Solvated docking protocol with explicit interfacial water molecules for improved accuracy
Clustering, Scoring & Ranking
Energy-based clustering to identify highly populated clusters of low-energy conformations
Selection of representative structures from each cluster for detailed analysis
Interface analysis including contact maps, hydrogen bond networks, and electrostatic complementarity
Support for modified nucleotide assessment in binding interface evaluation
Flexible Refinement & Delivery
Local flexible refinement of both protein side chains and nucleic acid backbone/sugar conformations
Energy minimization of top-ranked representative structures
Molecular dynamics-based stability assessment of predicted complexes
Comprehensive report with structural models, interaction analysis, and visualization-ready files
Dedicated Statistical Potentials: Our scoring functions are specifically parameterized for assessing protein–nucleic acid interactions, with separate statistical potentials for protein–DNA and protein–RNA complexes, ensuring accurate discrimination of near-native binding modes.
Solvated Docking Protocol: We incorporate explicit interfacial water molecules into the docking procedure, capturing the critical role of solvent-mediated contacts that often stabilize protein–nucleic acid interfaces.
Multi-Level Restraint Integration: Our platform supports filtering of output predictions based on blocking residues, binding site residues, and pairwise distance restraints, ensuring that experimentally derived knowledge is fully leveraged.
Robust Clustering and Ranking: We apply advanced clustering algorithms to identify highly populated clusters of low-energy conformations, selecting representative models that reflect the most probable binding geometries.
Comprehensive Molecular Support: Our service handles modified amino acids, chemically modified DNA/RNA molecules, and hybrid DNA–RNA duplexes (e.g., dsRNA/DNA hybrids), covering the full range of biologically relevant nucleic acid substrates.
Case 1: Modeling a Transcription Factor–DNA Complex
Background:
A research group studying gene regulation required structural insight into a zinc finger transcription factor binding to its cognate promoter DNA sequence. Experimental efforts to crystallize the full complex had been unsuccessful due to conformational flexibility in the linker regions.
Solution:
Using Profacgen's protein–nucleic acid docking platform, we performed a rigid-body global search guided by the known DNA recognition code of the zinc finger domains. Base-specific contact restraints derived from sequence conservation analysis were incorporated to narrow the search space. The solvated docking protocol captured water-mediated hydrogen bonds at the protein–DNA interface that had been predicted by mutagenesis studies.
Results:
Docking identified key base-specific contacts in the major groove of the DNA, with the zinc finger motifs inserting into the major groove in a manner consistent with canonical recognition patterns. The model guided the design of engineered zinc finger proteins with altered sequence specificity, and subsequent experimental validation confirmed the predicted binding preferences with high accuracy.
Case 2: Characterizing a Programmable Nuclease Off-Target Interaction
Background:
A biotechnology company developing gene editing therapies needed to assess potential off-target cleavage at genomic loci with sequence mismatches relative to the designed guide RNA. Understanding the structural basis of mismatch tolerance was essential for optimizing guide RNA specificity.
Solution:
We modeled the programmable nuclease–sgRNA complex in complex with a series of mismatched DNA target sequences. Docking simulations incorporated the protein–DNA and RNA–DNA interfaces simultaneously, with the hybrid DNA–RNA duplex support of our platform enabling accurate representation of the R-loop structure. Local flexible refinement of both the nuclease domains and the nucleic acid components captured conformational adjustments at mismatch sites.
Results:
The predicted binding energy differences across the panel of mismatched targets showed strong correlation with experimentally measured cleavage efficiency. Structural analysis revealed that mismatches positioned within the seed region of the guide RNA induced greater conformational distortion than distal mismatches, consistent with the known seed-region sensitivity of programmable nucleases. These findings directly supported the optimization of guide RNA design to minimize off-target activity.
Q: What types of nucleic acids can be modeled in the docking simulations?
A: Our platform supports dsDNA (B-, A-, and Z-forms), ssDNA, dsRNA, ssRNA, and DNA–RNA hybrids. We also accommodate modified nucleotides (e.g., methylated bases, phosphorothioate, LNA) and non-standard base pairs when parameter files are available.
Q: What input information is required to initiate a docking project?
A: Minimum input: protein sequence/structure and nucleic acid sequence/structure. Accuracy improves with binding site residues, mutagenesis, cross-linking, footprinting, or related complex structures. If no structure is available, we perform homology modeling or ab initio prediction.
Q: How does the solvated docking protocol improve prediction accuracy?
A: Water molecules often bridge key hydrogen bonds at protein–nucleic acid interfaces. Our protocol explicitly places and optimizes interfacial waters during scoring and refinement, improving binding geometry prediction and discrimination of near-native complexes.
Q: Can conformational changes in the protein or nucleic acid be modeled?
A: Yes. We start with a rigid-body global search, followed by local flexible refinement of protein side chains and nucleic acid backbone/sugar moieties. For larger conformational changes, ensemble docking using multiple starting conformations is available.
Q: What deliverables are provided upon completion of the project?
A: Deliverables include a detailed methodology report, ranked complex structures in PDB format, interface analysis (contact maps, hydrogen bonds, electrostatics), molecular dynamics stability assessment, publication-quality figures, and optional raw trajectories.
Q: How are the docking results validated, and what is the expected accuracy?
A: We validate via benchmarking, cluster analysis, and scoring consistency. When experimental data are available, we cross-validate predicted contacts. With binding site restraints and moderate conformational changes, near-native accuracy (interface RMSD < 3–4 Å) is achievable. Confidence metrics are transparently reported.
References:
Rodríguez-Lumbreras LA, Jiménez-García B, Giménez-Santamarina S, Fernández-Recio J. Pydockdna: a new web server for energy-based protein-dna docking and scoring. Front Mol Biosci. 2022;9:988996. doi:10.3389/fmolb.2022.988996
Online Inquiry
Fill out this form and one of our experts will respond to you within one business day.