At Profacgen, our Fold Recognition services deliver accurate structural fold identification and remote homology detection for proteins with low sequence identity to known structures, supporting structure prediction, functional annotation, and target discovery through advanced threading algorithms.
Protein tertiary structure is essential for understanding function and guiding drug discovery. Computational prediction serves as a powerful complement to experimental methods. Fold recognition (or threading) identifies structurally similar proteins to serve as templates for modeling, particularly when sequence identity falls below 25%.
Profacgen combines sequence profile-profile alignment with structural information to recognize correct folds. Our workflow involves building a template database, aligning the query sequence against each template via optimized scoring functions, and iterating across all known structures to find the best match. The most statistically probable alignment is used to construct a structural model by mapping backbone atoms onto the selected template. Unlike sequence-only approaches, our method leverages 3D structural data for enhanced accuracy. All models are quality-validated and suitable for downstream applications such as protein engineering and drug design.
Fold recognition (threading) addresses the challenge of predicting protein structures when sequence homology is insufficient for conventional comparative modeling:
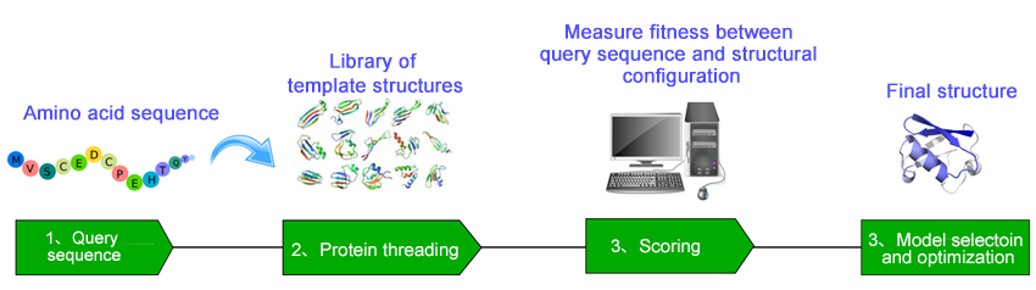 Figure 1. Fold recognition workflow: from template database construction and profile-profile alignment through scoring function optimization to structural model generation.
Figure 1. Fold recognition workflow: from template database construction and profile-profile alignment through scoring function optimization to structural model generation.
The output includes information about template proteins, the query-template alignments and most importantly full-length models of the target protein. We can also customize the service according to the specific requirements from our customers and integrate our computational procedures into your workflow.
Our fold recognition platform encompasses four specialized service modules, each addressing critical aspects of remote homology detection and structure prediction:
Fold Classification
Systematic assignment of query proteins to known structural fold classes.
Remote Homology Analysis
Detection of evolutionary relationships beyond sequence similarity thresholds.
Template Identification
Optimal template selection for downstream structure prediction and modeling.
Functional Annotation Support
Structure-based inference of molecular function from predicted folds.
Our Fold Recognition services support a broad spectrum of applications across structural biology and genomics:
Profacgen provides structured, analysis-ready documentation aligned with your fold recognition and structure prediction requirements:
| Deliverable | Description |
|---|---|
| Fold Recognition Reports | Comprehensive documentation of fold assignments, confidence scores, statistical significance, structural class annotations, and comparison to known fold families with evolutionary analysis |
| Candidate Structural Templates | Ranked list of optimal template structures with alignment details, coverage maps, scoring function values, and energy validation results for downstream modeling |
| Functional Annotation Results | Predicted molecular functions, active site architectures, binding pocket characteristics, and pathway associations derived from structural fold similarity and template functional annotations |
Program Context:
A microbiology research group identified a family of 200 uncharacterized proteins from a deep-sea metagenomic dataset. Standard BLAST searches returned no significant hits, preventing functional and structural annotation.
Objective:
To assign structural folds and predict functions for the protein family using fold recognition, enabling experimental prioritization and mechanistic hypothesis generation.
Approach:
Profacgen constructed sequence profiles for the protein family and performed threading against a comprehensive template database of 50,000 known folds. Profile-profile alignment and 1D-3D scoring identified a conserved α/β hydrolase fold with significant confidence. Multiple threading methods converged on the same fold assignment. Functional annotation based on the template active site architecture predicted esterase activity.
Outcome:
Fold recognition assigned the α/β hydrolase fold to 85% of the family members with high confidence. Experimental testing of 10 representative proteins confirmed esterase activity in 8, validating the structure-based functional prediction and enabling focused biochemical characterization of the family.
Program Context:
A pharmaceutical company identified a kinase-like protein as a potential oncology target but could not find suitable templates for homology modeling using standard sequence-based searches, as the closest homologues shared only 18% sequence identity.
Objective:
To identify structurally suitable templates for homology modeling and assess the feasibility of structure-based drug design despite low sequence homology.
Approach:
Profacgen performed fold recognition using profile-profile alignment and structural feature comparison. Threading identified a protein kinase fold with significant statistical confidence despite the low sequence identity. The optimal template was selected based on scoring function optimization and energy validation. A structural model was constructed and refined for ATP-binding pocket analysis.
Outcome:
The fold recognition approach identified a suitable kinase template that sequence methods missed. The resulting model revealed a druggable ATP-binding pocket with conserved hinge region geometry, enabling structure-based virtual screening. Two hit compounds from the screen showed micromolar activity in biochemical assays, validating the template and demonstrating the value of threading for challenging targets.
Fill out this form and one of our experts will respond to you within one business day.