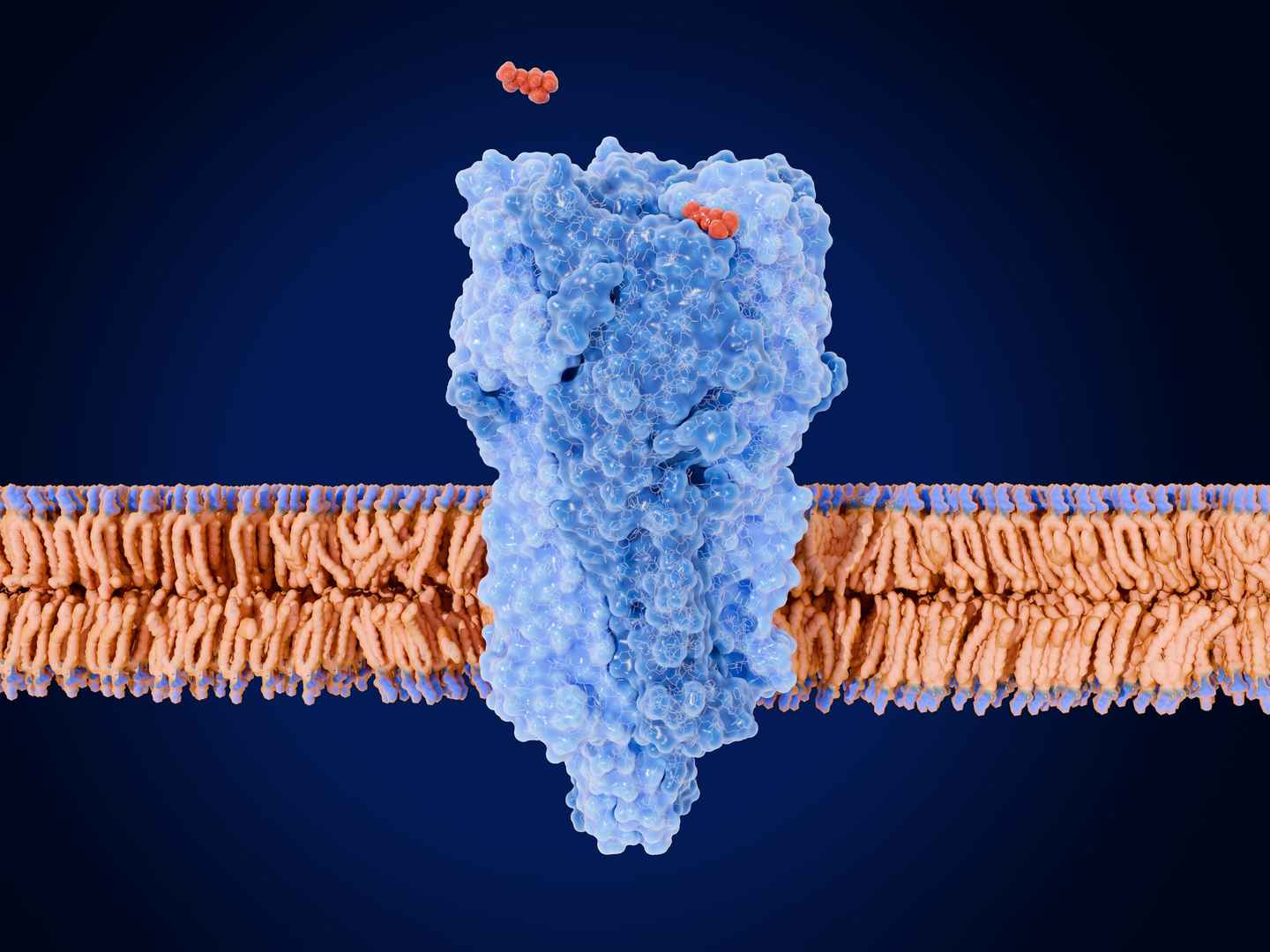
Profacgen offers Membrane Protein Modeling services, providing high-quality 3D structural models of GPCRs, ion channels, and transporters to support drug target characterization, ligand binding analysis, and structure-based design through advanced computational approaches.
Membrane proteins account for one-third of the human genome and over 50% of current drug targets, yet experimental structures remain scarce due to expression and reconstitution challenges. Computational modeling offers a valuable alternative for atomic-level structure elucidation. Our approach follows a homology-based workflow: template selection via sequence alignment, core structure prediction and refinement of membrane-spanning regions, followed by loop conformation modeling—particularly for extra-membranous domains—to generate complete models.
Profacgen enhances conventional homology modeling with membrane-protein-specific adaptations to avoid soluble-protein bias. When close homologs are unavailable, we exploit distant structural templates for core and loop reconstruction. With extensive experience in GPCR and transporter modeling, we deliver quality-verified structures suitable for computational drug design, molecular dynamics simulation, and structure-based protein engineering.
Membrane protein structure prediction addresses unique challenges that distinguish these targets from soluble proteins:
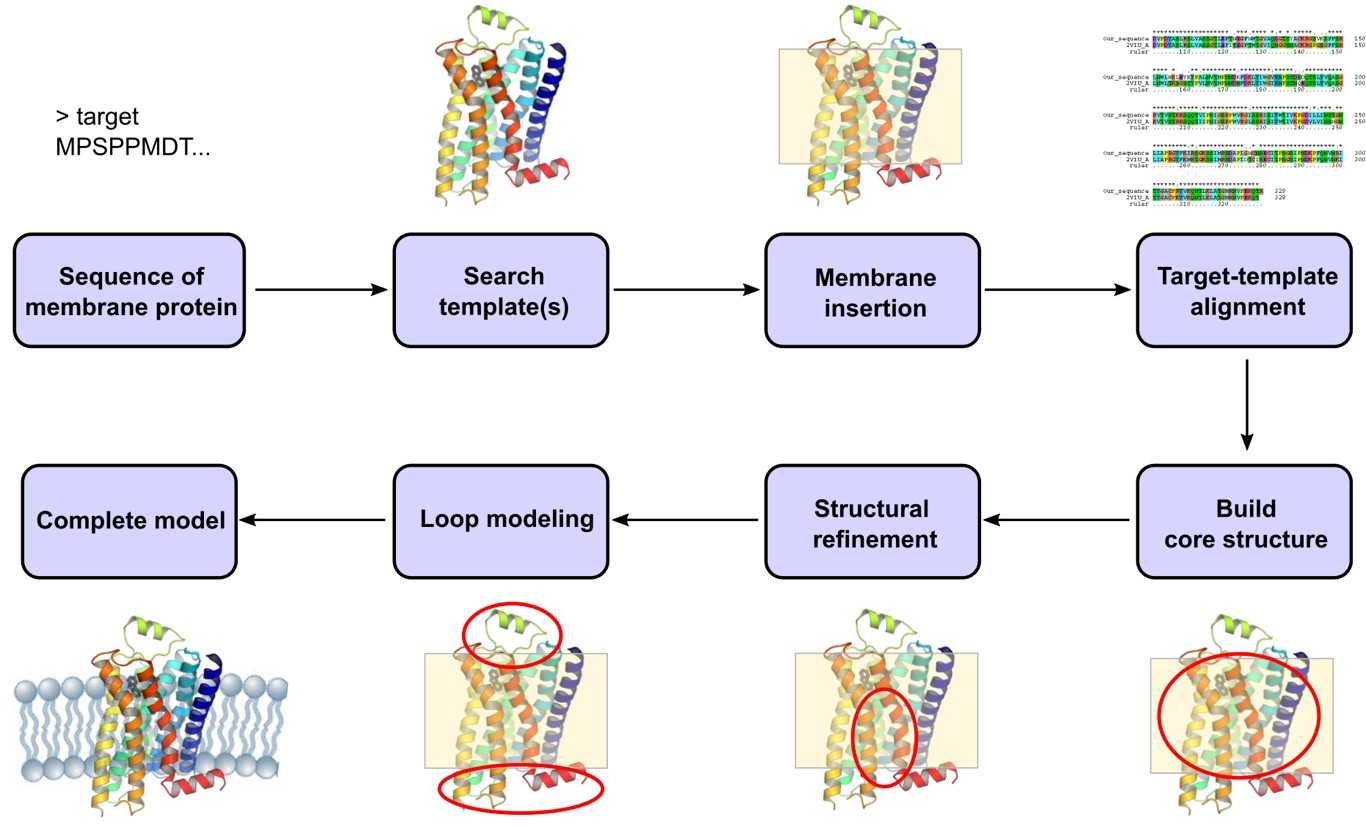 Figure 1. Membrane protein modeling pipeline: from sequence alignment and transmembrane region prediction through core construction, loop modeling, and membrane embedding.
Figure 1. Membrane protein modeling pipeline: from sequence alignment and transmembrane region prediction through core construction, loop modeling, and membrane embedding.
Our goal is to predict the structure of membrane proteins from their amino acid sequences with an accuracy comparable to experimental approaches, overcoming many difficulties in the structure determination of membrane proteins.
Our membrane protein modeling platform encompasses four specialized service modules, each optimized for distinct membrane protein classes and analytical objectives:
GPCR Modeling
Structure prediction and refinement for G protein-coupled receptors, the largest and most pharmaceutically important membrane protein family.
Ion Channel Modeling
Accurate structural prediction of channel architectures and pore geometries for mechanistic and pharmacological studies.
Transporter Modeling
Structural modeling of substrate translocation pathways and alternating access mechanisms.
Membrane Environment Refinement
Embedding and equilibration of membrane protein models in physiologically relevant lipid bilayers.
Our Membrane Protein Modeling services support a broad spectrum of applications across pharmaceutical and biotechnological development:
Profacgen provides structured, analysis-ready documentation aligned with your membrane protein modeling and drug discovery requirements:
| Deliverable | Description |
|---|---|
| Refined Membrane Protein Models | PDB-format coordinate files for complete membrane protein models, including transmembrane domains, loops, and membrane-embedded coordinates with lipid bilayer positioning |
| Structural Evaluation Reports | Quality assessment metrics including transmembrane topology validation, helix packing scores, loop geometry analysis, and membrane insertion energy profiles |
| Binding Site Analysis | Orthosteric and allosteric pocket identification, residue-level mapping of ligand-accessible regions, pocket volume and druggability scores, and electrostatic surface analysis |
Program Context:
A pharmaceutical company required a structural model of a Class A GPCR target for which no experimental structure was available, to support a virtual screening campaign for novel antagonists. The target shared only moderate sequence identity with existing GPCR templates.
Objective:
To generate a high-quality GPCR structural model suitable for structure-based virtual screening, including accurate transmembrane helix packing and extracellular loop conformations for ligand binding pocket definition.
Approach:
Profacgen performed HMM-based template recognition across the GPCR structural database, identifying a distant homologue with conserved transmembrane topology. The seven-helix bundle core was constructed and refined using membrane-specific packing algorithms. Extracellular loops were modeled iteratively, and the model was embedded in a POPC lipid bilayer followed by molecular dynamics equilibration. The orthosteric binding pocket was validated against known ligand pharmacophores.
Outcome:
The refined GPCR model achieved high topology confidence scores and was successfully used for virtual screening of 2 million compounds, yielding 150 prioritized hits. Experimental validation of 20 selected compounds confirmed 8 with micromolar affinity, validating the model's utility for drug discovery.
Program Context:
A research group studying an MFS transporter required structural models of both outward-facing and inward-facing conformations to understand the substrate translocation mechanism and identify residues critical for alternating access.
Objective:
To generate conformational state-specific models of the transporter and identify the substrate translocation pathway, binding residues, and conformational switch mechanisms.
Approach:
Profacgen selected outward-facing and inward-facing templates from available MFS structures, performed sequence alignment with attention to transmembrane helix boundaries, and constructed core models for each state. Loop regions were modeled to close the extracellular and intracellular gates appropriately. Molecular dynamics simulations were performed to assess state stability, and substrate docking identified the central binding pocket and translocation pathway residues.
Outcome:
The two conformational models revealed a rocker-switch mechanism involving helices TM1-TM2 and TM7-TM8, with 12 conserved residues forming the substrate translocation pathway. Mutagenesis studies guided by the models confirmed 3 critical residues for substrate binding, validating the mechanistic insights.
Fill out this form and one of our experts will respond to you within one business day.