We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our
Privacy Policy
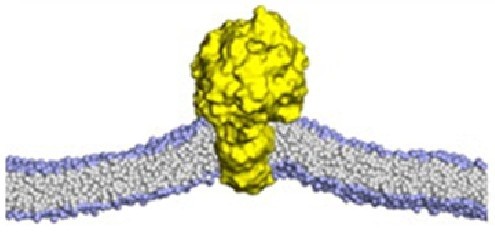
Membrane proteins are central to numerous cellular functions, and their association with lipids regulates signaling and key biological events. Lipids stabilize protein structures, drive conformational rearrangements, and serve as substrates for metabolic enzymes. Cytosolic proteins are often recruited to membranes via specific lipid-binding domains.
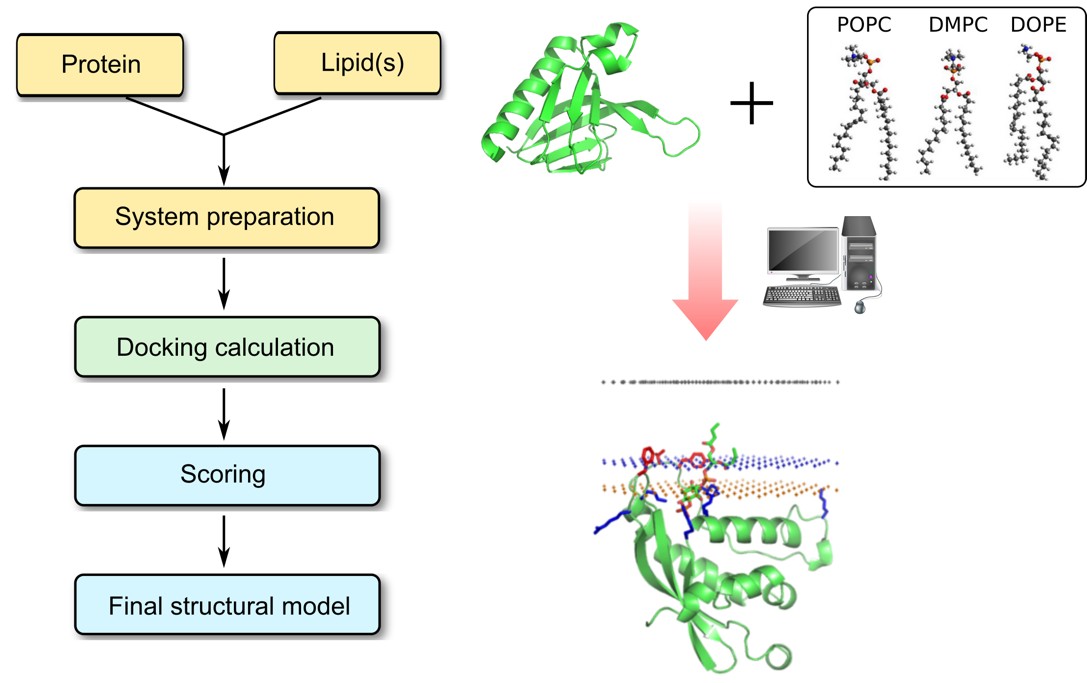
Protein–lipid interactions are governed by hydrogen bonds, van der Waals forces, hydrophobic contacts, and ionic bridges—with Asp and Glu residues playing key roles in electrostatic anchoring. Profacgen offers Protein–Lipid Docking service, using advanced computational tools to predict these interactions at atomic resolution. Binding sites are derived from sequence or structure via molecular docking, using a scoring function tailored to lipid distortion and protein flexibility. Experimental restraints can be applied to narrow the search, and top poses are clustered and validated through MD simulations. This integrated approach enables precise identification of lipid-binding regions and elucidation of the physicochemical drivers of protein–lipid recognition.
Why Protein–Lipid Docking?
Understanding how proteins interact with membrane lipids is fundamental to elucidating the molecular mechanisms underlying a vast array of biological processes. Protein–lipid interactions govern everything from membrane protein folding and stability to signal transduction, vesicle trafficking, and enzyme catalysis. Despite their importance, these interactions remain notoriously difficult to characterize experimentally due to the dynamic and heterogeneous nature of biological membranes.
Computational protein–lipid docking offers a powerful complement to experimental techniques by providing atomic-level insights into:
The precise location and geometry of lipid binding sites on protein surfaces
The physicochemical forces driving specific lipid recognition, including hydrogen bonding networks, hydrophobic packing, and electrostatic complementarity
The conformational changes induced in both protein and lipid upon complex formation
The energetic landscape of protein–lipid association, enabling comparative analysis of different lipid species
The molecular basis for lipid selectivity and specificity across related protein families
These insights are invaluable for structure-based drug design targeting membrane proteins, engineering proteins with tailored lipid-binding properties, and understanding disease mechanisms arising from aberrant protein–lipid interactions.
Protein–Lipid docking process
Our Protein–Lipid Docking Service Offerings
Services
Details
Membrane Protein & Receptor Preparation
Transmembrane domain modeling and orientation prediction
Membrane-embedded structure preparation with explicit or implicit bilayer representation
Binding site prediction using sequence conservation, structural features, and physicochemical properties
Protein protonation state assignment at membrane-relevant pH
Lipid Library Construction
Support for up to 32 different lipid types, including phospholipids, sphingolipids, sterols, and glycolipids
Custom lipid parameterization for non-standard or modified lipid species
Detergent and micelle support for membrane mimetic systems
Generation of diverse lipid conformer ensembles for flexible docking
Protein–Lipid Docking
Combined sequence- and structure-based docking approach for comprehensive binding site exploration
Lipid-specific scoring function designed to capture hydrophobic matching, headgroup interactions, and acyl chain packing
Membrane distortion modeling to account for bilayer deformation upon protein–lipid binding
Incorporation of experiment-derived restraints to guide and refine docking predictions
Clustering, Scoring & Selection
Binding energy-based clustering to identify representative binding modes
Lipid distortion analysis to evaluate conformational strain in bound lipids
Protein conformational change assessment to capture induced-fit effects
Ranking of top-scoring complexes with detailed interaction fingerprints
MD Refinement & Delivery
Explicit solvent molecular dynamics simulations for stability testing of predicted complexes
Interaction interface analysis including hydrogen bond occupancy, contact maps, and energy decomposition
Comparison of binding free energies across different lipid types and binding poses
Comprehensive report with visualizations, structural coordinates, and actionable recommendations
Dedicated Scoring Functions: Our platform employs statistical potentials specifically designed and parameterized to assess protein–lipid interactions, accounting for the unique physicochemical environment of the membrane–water interface.
Comprehensive Binding Mechanism Coverage: We support the full spectrum of membrane binding mechanisms, including covalent attachment, hydrophobic insertion, electrostatic interactions, hydrogen bonding networks, and van der Waals packing.
Extensive Lipid Library: Our service supports up to 32 different lipid types with appropriate selection of lipid type and composition, enabling systematic screening of lipid specificity and selectivity.
Membrane Environment Modeling: We explicitly simulate the effect of protein–lipid binding on membrane distortion and protein structural change, providing a realistic representation of the bilayer environment. The platform is also customizable to support single detergent molecules or micelles.
Seamless MD Integration: All docking results are designed for seamless integration with downstream molecular dynamics simulations, enabling rigorous validation and refinement of predicted complexes under physiologically relevant conditions.
G protein-coupled receptors (GPCRs) are embedded in the plasma membrane and their structure, dynamics, and function are profoundly influenced by surrounding lipids. A client sought to identify specific cholesterol and phospholipid interaction sites on their target GPCR to guide stabilization strategies for structural biology efforts.
Solution:
We performed systematic protein–lipid docking against the GPCR structure, screening a focused lipid library including cholesterol and key phospholipid species. The docking protocol incorporated sequence conservation analysis to prioritize functionally relevant binding regions, and the top-scoring complexes were subjected to explicit membrane MD simulations for validation.
Results:
The docking calculations revealed multiple conserved cholesterol binding motifs on the receptor surface, including a high-occupancy site at the TM4–TM5 interface consistent with known GPCR–cholesterol interaction patterns. The identified lipid interaction map directly guided the design of a stabilization strategy that facilitated successful crystallization of the receptor.
Case 2: Studying PIP₂-Mediated Membrane Targeting of a PH Domain
Background:
Pleckstrin homology (PH) domains mediate membrane recruitment of signaling proteins through specific recognition of phosphoinositide lipids. A client needed to determine the molecular basis for phosphoinositide binding specificity in a novel PH domain implicated in cancer cell migration.
Solution:
We modeled the PH domain structure and performed comparative docking of PI(4,5)P2 and PI(3,4)P2 at the canonical binding pocket. The docking protocol explicitly accounted for the membrane surface, lipid headgroup flexibility, and electrostatic interactions. Binding free energies were calculated and compared between the two phosphoinositide species.
Results:
The docking analysis predicted a clear energetic preference for PI(4,5)P2 over PI(3,4)P2, with key specificity determinants identified at the 4- and 5-phosphate positions. These predictions matched subsequent surface plasmon resonance (SPR) binding data with high fidelity. Furthermore, the calculations elucidated the electrostatic switch mechanism by which phosphorylation-dependent charge changes modulate membrane association, providing mechanistic insight into the domain's role in cell migration signaling.
Q: What types of lipid molecules can Profacgen's docking platform handle?
A: Our platform supports 32 lipid classes, including phospholipids (PC, PS, PE, PI and phosphoinositides), sphingolipids (sphingomyelin, ceramide, gangliosides), sterols (cholesterol, ergosterol), and glycolipids. Custom parameterization for non-standard lipids, detergents, and micelle systems is also available.
Q: Do I need an experimental structure of my protein to use this service?
A: No. If available, we use crystal or cryo-EM structures directly. If not, we generate high-quality homology models including transmembrane domains. Our protocol integrates both sequence and structure information, so even without a full 3D structure, sequence-based binding site predictions provide useful starting points.
Q: How does the docking protocol account for membrane distortion and protein flexibility?
A: Our scoring function accounts for lipid distortion and protein conformational changes, with explicit membrane representation including bilayer deformation (thickness, curvature). Protein side-chain flexibility is sampled, and larger changes are assessed via clustering. Final refinement via MD in explicit membrane validates binding modes under physiological conditions.
Q: What experimental data can be incorporated to improve docking accuracy?
A: We incorporate mutagenesis, cross-linking MS, NMR chemical shift perturbation, and functional specificity data to constrain and direct the docking search toward biologically relevant solutions, improving prediction accuracy.
Q: How are the final binding modes validated and what deliverables can I expect?
A: Top poses undergo explicit-solvent MD to assess stability, with analysis of H-bond occupancy, contact persistence, and binding free energies. Deliverables include a full methodology report, PDB coordinates of predicted complexes, publication-quality figures, and interaction analysis (per-residue energy decomposition, contact summaries).
Q: Can Profacgen compare binding preferences across multiple lipid species in a single project?
A: Yes. We systematically dock up to 32 lipid species against your target and rank them by predicted affinity, revealing selectivity patterns tied to headgroup, acyl chain, or saturation. This identifies allosteric sites and guides experimental focus toward the most relevant lipid partners.
References:
Corradi V, Sejdiu BI, Mesa-Galloso H, et al. Emerging diversity in lipid–protein interactions. Chem Rev. 2019;119(9):5775-5848. doi:10.1021/acs.chemrev.8b00451
Online Inquiry
Fill out this form and one of our experts will respond to you within one business day.