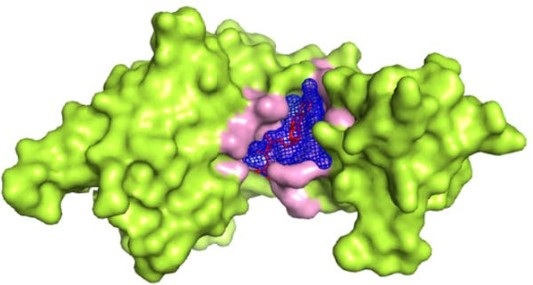
The interaction between proteins and their cognate ligands plays an essential role in numerous biological processes and metabolic pathways, including signal transduction, molecular transport, cell regulation, gene expression control, and enzyme inhibition. Understanding these interactions at the atomic level is fundamental to elucidating disease mechanisms and developing novel therapeutic agents. Profacgen's Protein–Ligand Docking service leverages state-of-the-art computational methodologies to predict the preferred binding pose and affinity of small-molecule ligands within protein receptor binding sites, providing researchers with critical structural insights that guide rational drug design and mechanistic studies.
While experimental techniques such as X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy remain the gold standards for determining protein–ligand complex structures, they are often constrained by significant limitations — including the difficulty of obtaining diffraction-quality crystals, the size restrictions of NMR, and the considerable time and cost involved. Computational docking has therefore emerged as a powerful and complementary approach for studying protein–ligand interactions at scale. Profacgen's platform integrates multiple validated docking engines with advanced scoring functions to deliver accurate, reproducible, and biologically meaningful results tailored to the specific needs of each project.
Protein–ligand docking serves as a cornerstone technique in modern structure-based drug discovery. By computationally simulating the interaction between a protein receptor and a small-molecule ligand, researchers can rapidly evaluate binding hypotheses, prioritize compounds for synthesis, and gain mechanistic understanding without the bottlenecks associated with purely experimental approaches. Every docking protocol can be understood as a combination of two core components: a search algorithm that explores the conformational and positional space of the ligand, and a scoring function that estimates the binding affinity of each generated pose. The synergy between efficient sampling and accurate energy evaluation determines the reliability of the predictions.
Profacgen's docking workflow mimics the natural course of ligand–receptor association by identifying the lowest-energy pathway for binding. This physics-based approach ensures that the predicted poses are not merely geometrically plausible but also energetically favorable under physiological conditions. The applications of protein–ligand docking span the full breadth of drug discovery and chemical biology:
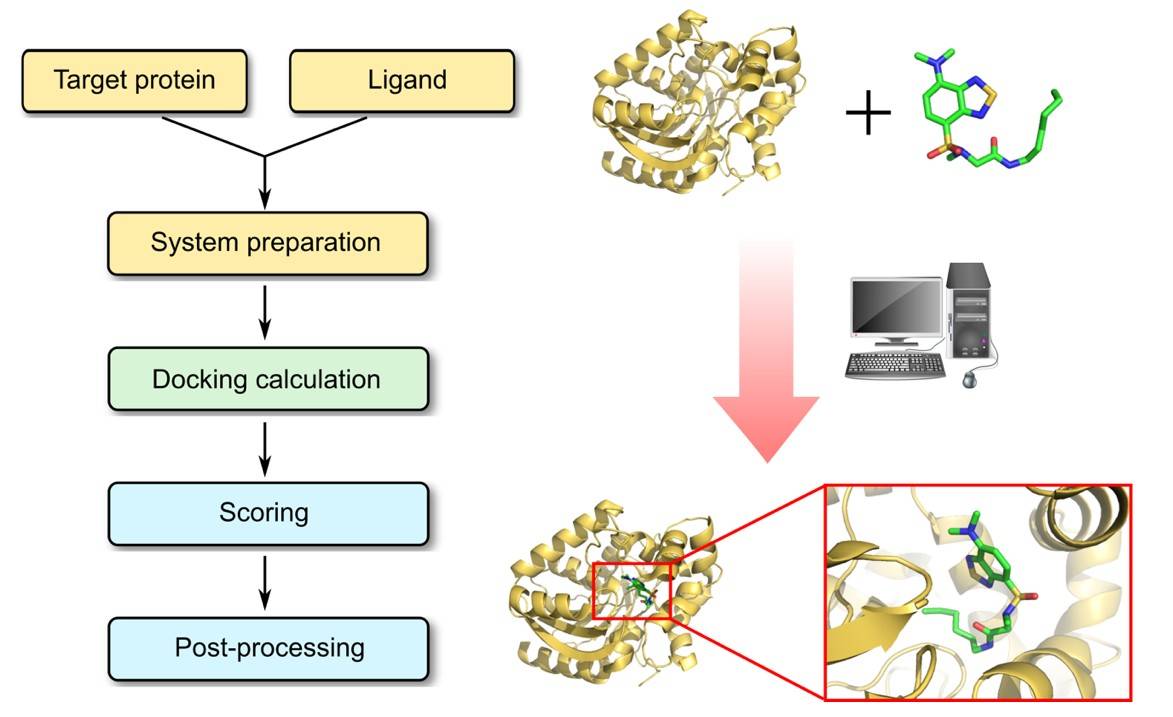
Profacgen provides a comprehensive suite of protein–ligand docking services designed to address the diverse requirements of academic research groups, biotechnology companies, and pharmaceutical organizations. Our modular service structure allows clients to engage individual components or the complete workflow depending on project needs.
| Service Component | Description |
|---|---|
| Target Preparation & Binding Site Definition |
|
| Ligand Preparation & Library Generation |
|
| Docking & Pose Generation |
|
| Scoring & Ranking |
|
| Post-Docking Analysis & Reporting |
|
Background:
A biotechnology company was developing a series of type II kinase inhibitors targeting an oncology indication. Despite promising biochemical potency, the team lacked structural understanding of the binding modes adopted by their lead compounds, which hindered rational medicinal chemistry efforts to improve selectivity over closely related off-target kinases. Experimental co-crystallization attempts had been unsuccessful due to poor diffraction quality.
Our Solution:
Profacgen employed an induced-fit docking protocol that permitted conformational adaptation of both the kinase hinge region and the DFG-out activation loop to accommodate the type II inhibitor scaffold. Docking was performed against the target kinase structure as well as a panel of 15 related kinases to evaluate selectivity determinants. Consensus scoring with MM/GBSA rescoring was applied to rank binding poses, and per-residue interaction energy decomposition identified the key residue-level contacts governing target affinity versus off-target binding.
Final Results:
The docking analysis revealed that a single hinge-region hydrogen bond mediated by a backbone NH group was the primary determinant of target engagement, while steric clashes with a bulkier residue in the off-target kinases explained selectivity. Guided by these structural insights, the client's medicinal chemistry team introduced modifications that preserved the critical hinge interaction while exacerbating the steric penalty at off-targets, ultimately achieving a 50-fold improvement in selectivity with maintained nanomolar potency.
Background:
A mid-sized pharmaceutical company sought to identify novel chemical starting points for a class A G protein-coupled receptor (GPCR) target implicated in metabolic disease. The target lacked known drug-like ligands, and the company required a diverse set of validated hit compounds to initiate a lead discovery program. A virtual screening campaign was commissioned to evaluate approximately 2 million commercially available compounds from multiple vendor libraries.
Our Solution:
Profacgen constructed a high-quality receptor model using the available cryo-EM structure, with careful attention to the protonation states of key residues in the orthosteric binding pocket. The compound library was prepared with exhaustive enumeration of tautomers, stereoisomers, and protonation states at physiological pH. A tiered docking strategy was implemented: an initial high-throughput rigid docking stage to triage the full library, followed by flexible docking with a refined scoring function for the top 50,000 compounds. Consensus scoring across three orthogonal scoring functions was used to select the final 50 compounds for experimental validation.
Final Results:
Of the 50 compounds selected for biochemical testing, 12 demonstrated confirmed activity in the primary assay, yielding a hit rate of 24% — substantially exceeding industry benchmarks for virtual screening campaigns. Three compounds exhibited sub-micromolar affinity (IC50 values of 180 nM, 420 nM, and 760 nM) with favorable ligand efficiency metrics. The hits represented four distinct chemotypes, providing multiple structural starting points for the client's medicinal chemistry optimization program.
References:
Fill out this form and one of our experts will respond to you within one business day.