The research and development of therapeutic drugs is an expensive and time-consuming process. So far, many scientific methods have been applied for drug design, and one of the most popular approaches is computer-aided drug design (CADD). Taking advantage of computational techniques, the modeling of quantitative structure-activity relationships (QSAR) explores the relationship between chemical structures and biological activity of a given set of small molecules. The resulting QSAR models can be used to predict the activities of new chemicals, including affinities of ligands to their binding sites, inhibition constants, rate constants and others, based on certain structural features or with atomic, group or molecular properties, such as lipophilicity, polarizability, electronic and steric properties. As an extension to the classical QSAR approaches pioneered by Hansch and Free-Wilson, 3D-QSAR exploits the three-dimensional properties of the ligands to predict their biological activities using robust chemometric techniques such as PLS, G/PLS and ANN etc. This method can effectively guide the selection of promising compounds that need to be synthesized and predict the activity of those compounds leading to the structural optimization.
Profacgen provides 3D-QSAR pipeline that enables researchers to effectively perform force field calculations requiring three-dimensional structures of a training set (with known activities measured experimentally). The created data space is then reduced by feature extraction and a following machine learning method. Our workflow includes biological data analysis, optimization of 3D structure of the biomolecules, determination of bioactive conformations of biomolecules, calculation of molecular interaction energy fields, 3D QSAR model generation and validation, etc. This one-step 3D-QSAR workflow aims to help researchers identify determinants for biological activities of small molecules, optimize existing leads for improved activities, and predict the biological activities of untested compounds.
Structure- and Ligand-Based 3D QSAR Protocol
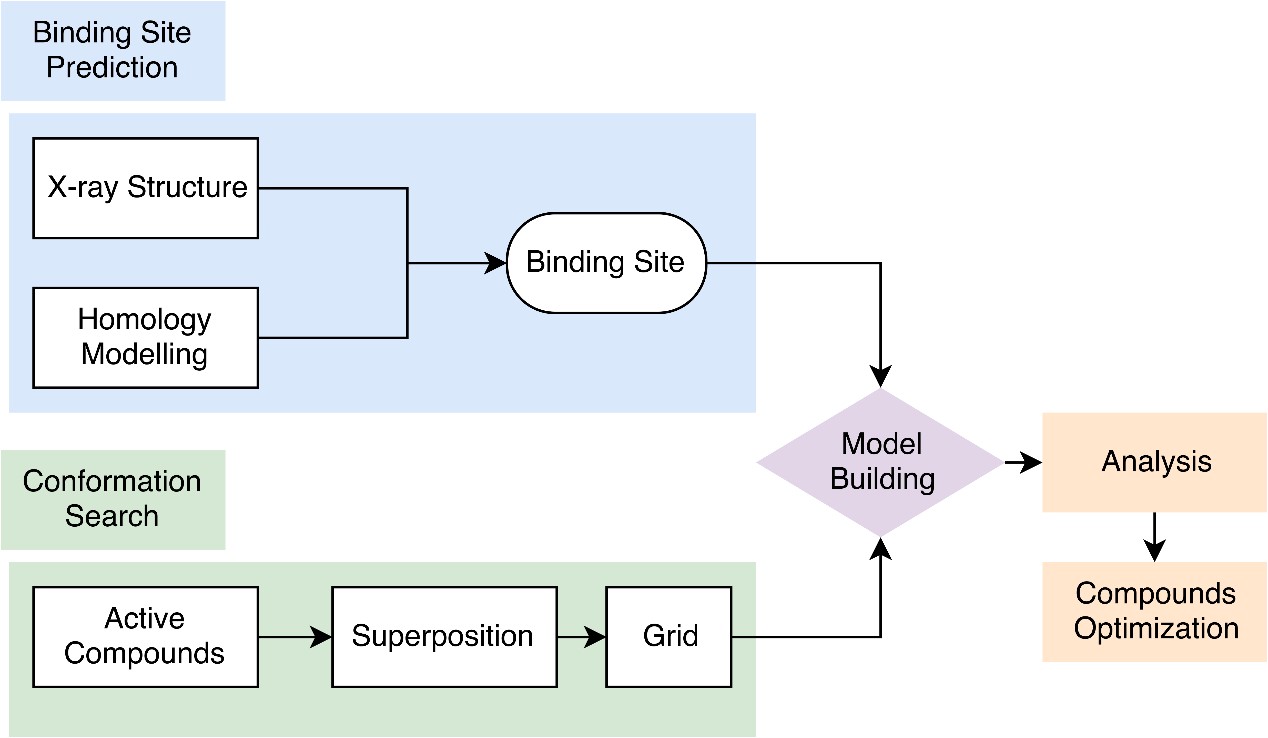
Our 3D-QSAR analysis represents a valuable medicinal chemistry tool, and the ever-increasing information from structural biology will surely present valuable feedback to the assumptions that form the basis of 3D-QSAR methods. The generated QSAR models will be thoroughly validated both internally as well as externally (in collaboration with the customers) using rigorous cross-validation techniques.
The 3D-QSAR protocol is highly customizable according to the specific requirements from the customers. Please do not hesitate to contact us for more details about our services.
Fill out this form and one of our experts will respond to you within one business day.