We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our
Privacy Policy
At Profacgen, our Homology Modeling services deliver high-quality 3D protein structures through template-based comparative modeling, supporting structure-based drug discovery, protein engineering, functional analysis, and mutation studies with accuracy approaching experimental methods.
Protein structure is key to function and drug design, yet experimental determination remains costly and slow. Homology modeling offers a reliable computational alternative, building atomic-resolution models from amino acid sequences using related homologs as templates—leveraging the fact that structure is more conserved than sequence.
Profacgen models diverse monomeric and oligomeric proteins, delivering quality-validated structures for gene annotation, molecular docking, and downstream engineering or design applications.
Overview of Homology Modeling
Figure 1. Homology modeling workflow: from template identification and sequence alignment through model construction, refinement, and validation.
Homology modeling is the most reliable computational approach for predicting protein structures when experimental data is unavailable:
Template-based structure prediction: Identification and utilization of one or more experimentally determined structures of related homologous proteins as templates for building atomic-resolution models of target proteins from sequence information
Sequence alignment: Accurate alignment of target and template sequences with manual corrections to ensure proper mapping of structurally conserved regions, loops, and functional motifs
Model construction: Assembly of three-dimensional coordinates for the target protein by copying conserved backbone regions and rebuilding variable regions using optimized geometric and energetic criteria
Structural refinement: Energy minimization, loop modeling, side-chain optimization, and validation against stereochemical and energetic quality metrics to ensure physical realism and accuracy
Our goal is to predict a structure of protein from its sequence with an accuracy comparable to the best results achieved experimentally. Large-scale automated modeling of protein-coding regions in a genome is also available at Profacgen.
Our Modeling Capabilities
Our homology modeling platform encompasses four interconnected service modules, each addressing a critical stage of the comparative modeling pipeline:
Template Identification
Systematic search and selection of optimal structural templates from curated databases.
Multiple-template recognition and selection from PDB and specialized structural databases
Sequence similarity assessment, structural quality evaluation, and phylogenetic analysis
Coverage optimization to maximize structurally resolved regions of the target sequence
Template combination strategies for multi-domain and chimeric protein targets
Sequence Alignment
Precision alignment of target and template sequences with structural awareness.
Accurate sequence alignment with manual corrections informed by secondary structure prediction
Profile-based and HMM-guided alignment for distantly related homologues
Loop region and insertion/deletion positioning with geometric constraints
Alignment validation through scoring functions and structural consistency checks
Model Construction
Assembly of three-dimensional coordinates with conserved and rebuilt regions.
Iterative loop modeling and side-chain optimization for variable regions
Prediction of multi-oligomeric proteins with subunit assembly and interface prediction
Support for induced fit effect upon binding of ligand or cofactor
Support for incorporation of unnatural amino acids and post-translational modifications
Structure Refinement and Validation
Optimization and quality assessment of final models using multiple criteria.
Energy minimization and molecular dynamics refinement for geometric optimization
Quality assessment by multiple criteria including Ramachandran analysis, clash scores, and energy profiles
Support for additional experimental restraints to improve model accuracy
Statistical confidence scoring and error estimation for model reliability assessment
Applications
Our Homology Modeling services support a broad spectrum of applications across structural biology and pharmaceutical development:
Structure-Based Drug Discovery: Generation of target protein structures for virtual screening, molecular docking, pharmacophore modeling, and lead optimization when experimental structures are unavailable
Protein Engineering: Structural guidance for rational design of improved enzymes, therapeutic proteins, and biologics with enhanced stability, activity, or specificity
Functional Analysis: Functional annotation of genes through structural characterization, active site identification, and prediction of molecular interactions and catalytic mechanisms
Mutation Studies: Structural interpretation of disease-associated mutations, polymorphism effects, and structure-function relationships through comparative modeling of variant proteins
Deliverables
Profacgen provides structured, analysis-ready documentation aligned with your structural modeling and discovery requirements:
Deliverable
Description
3D Protein Models
PDB-format coordinate files for monomeric, oligomeric, and ligand-bound protein models, including all atoms, secondary structure assignments, and chain identifiers
Validation Reports
Comprehensive quality assessment including Ramachandran statistics, clash scores, rotamer analysis, energy profiles, template coverage maps, and confidence scores for each model region
Structural Analysis
Secondary structure content, active site and binding pocket identification, electrostatic surface analysis, conservation mapping, and comparison to template structures with deviation metrics
Multiple-Template Expertise: We employ sophisticated multiple-template recognition and selection strategies, combining structural information from several homologues to maximize model accuracy and coverage.
Precision Alignment Protocols: Our accurate sequence alignment with manual corrections ensures proper mapping of structurally conserved regions, particularly critical for distantly related proteins with low sequence identity.
Advanced Refinement Methods: We perform iterative loop modeling and side-chain optimization, supported by energy minimization and molecular dynamics refinement to achieve physically realistic conformations.
Flexible Modeling Scope: We support prediction of multi-oligomeric proteins, induced fit effects upon ligand binding, additional experimental restraints, and incorporation of unnatural amino acids for specialized applications.
Scenario 1: Structure-Based Virtual Screening for an Orphan Target
Program Context:
A pharmaceutical company identified a novel kinase target through genomic screening but lacked experimental structural data. The target shared moderate sequence identity with several structurally characterized kinases, making it a candidate for homology modeling to support a virtual screening campaign.
Objective:
To generate a high-quality homology model of the target kinase suitable for structure-based virtual screening, including accurate ATP-binding pocket geometry and activation loop conformation for inhibitor design.
Approach:
Profacgen identified three kinase templates with complementary coverage of the target sequence and performed multiple-template alignment with manual correction of the activation loop region. The model was constructed with iterative loop modeling for the DFG motif and refined through molecular dynamics simulation. The ATP-binding pocket was validated by docking known kinase inhibitors and comparing poses to crystallographic complexes.
Outcome:
The refined model achieved high validation scores and was used for virtual screening of 1.5 million compounds. Experimental testing of 50 prioritized hits yielded 4 novel inhibitors with nanomolar affinity, validating the model's utility for drug discovery and providing structural insights for lead optimization.
Scenario 2: Mutation Impact Assessment for a Therapeutic Enzyme
Program Context:
A biotechnology company developed a therapeutic enzyme that showed reduced activity in a clinical variant. Understanding the structural basis of this loss-of-function mutation was critical for engineering a improved variant with restored activity.
Objective:
To generate structural models of wild-type and mutant enzyme, identify the structural consequences of the mutation, and guide rational design of compensatory mutations to restore catalytic efficiency.
Approach:
Profacgen built homology models of both wild-type and mutant enzyme using a closely related experimental structure as template. The models were refined and validated, followed by comparative analysis of active site geometry, hydrogen bond networks, and substrate binding poses. Molecular dynamics simulations revealed increased loop flexibility in the mutant near the catalytic site.
Outcome:
The structural analysis identified that the mutation disrupted a critical hydrogen bond stabilizing the catalytic loop. A compensatory double mutation was designed and modeled, predicted to restore the hydrogen bond network. Experimental validation confirmed 85% recovery of wild-type activity, demonstrating the value of structure-guided protein engineering.
Q: What is the minimum sequence identity required for reliable homology modeling?
A: Models based on templates with >30% sequence identity are generally reliable for backbone structure. Below 30%, accuracy decreases but our advanced methods including HMM-based alignment and multiple-template combination can still produce useful models for many applications.
Q: Can you model protein complexes and oligomers?
A: Yes. We predict multi-oligomeric proteins by modeling subunits individually and assembling them based on template complex structures or protein-protein docking, followed by interface refinement.
Q: Do you support ligand-bound or cofactor-bound models?
A: Yes. We support induced fit modeling upon ligand or cofactor binding. Known ligands can be incorporated during refinement to generate holo-protein models suitable for docking and mechanism studies.
Q: What validation metrics do you provide?
A: We provide Ramachandran plots, clash scores, rotamer analysis, energy profiles, template coverage maps, and statistical confidence scores. Each metric assesses different aspects of model quality.
Q: Can you incorporate experimental restraints into the model?
A: Yes. We support additional restraints from NMR, cross-linking, cryo-EM density, or mutagenesis data to improve model accuracy beyond template-based prediction alone.
Q: What input do you need to start a modeling project?
A: We require the amino acid sequence of the target protein. Additional information such as known ligands, experimental restraints, or preferred oligomeric state can improve model quality but is not mandatory.
Online Inquiry
Fill out this form and one of our experts will respond to you within one business day.
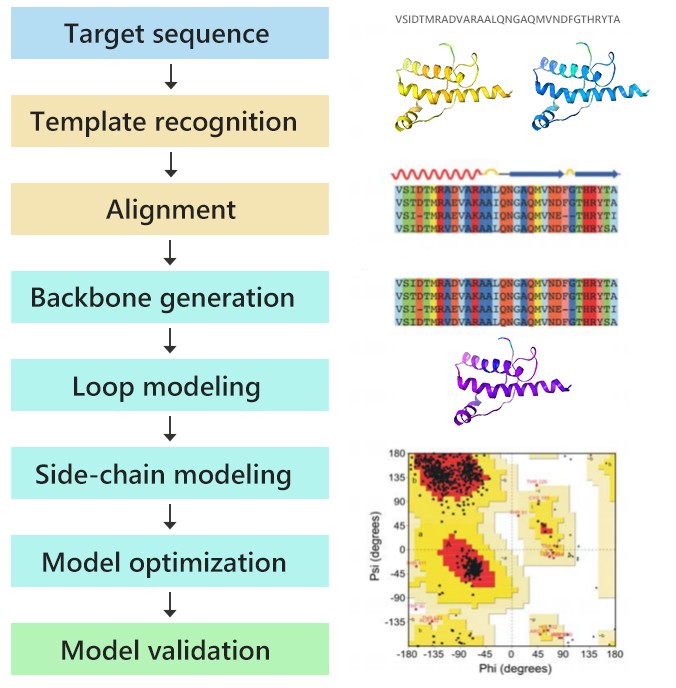 Figure 1. Homology modeling workflow: from template identification and sequence alignment through model construction, refinement, and validation.
Figure 1. Homology modeling workflow: from template identification and sequence alignment through model construction, refinement, and validation.