Profacgen offers Quantum Mechanics (QM)-Based Drug Design service, delivering high-accuracy computational insight into drug–receptor interactions by incorporating ab initio quantum chemistry into every stage of the computer-aided drug discovery (CADD) workflow. Drug discovery involves the identification of molecular candidates followed by the synthesis and testing of compounds for pharmaceutical efficacy. CADD uses computational methods to investigate and predict drug–receptor interactions, and QM has become an essential tool in CADD research, offering a physically rigorous description of electronic structure that empirical methods cannot match.
High-throughput in silico screening techniques such as docking and QSAR can significantly reduce the time required for compound discovery and optimization. However, these rapid methods often lack the accuracy needed to explore the details of binding mechanisms, especially when electronic effects, charge transfer, metal coordination, or covalent bond formation dominate the interaction. QM approaches obtain a more accurate representation of molecular systems by explicitly solving for the electronic structure of the molecules involved. Profacgen provides QM calculation services to describe the protein system—including ligands and solvent—so that clients gain a better understanding of protein–ligand interactions with markedly improved accuracy.
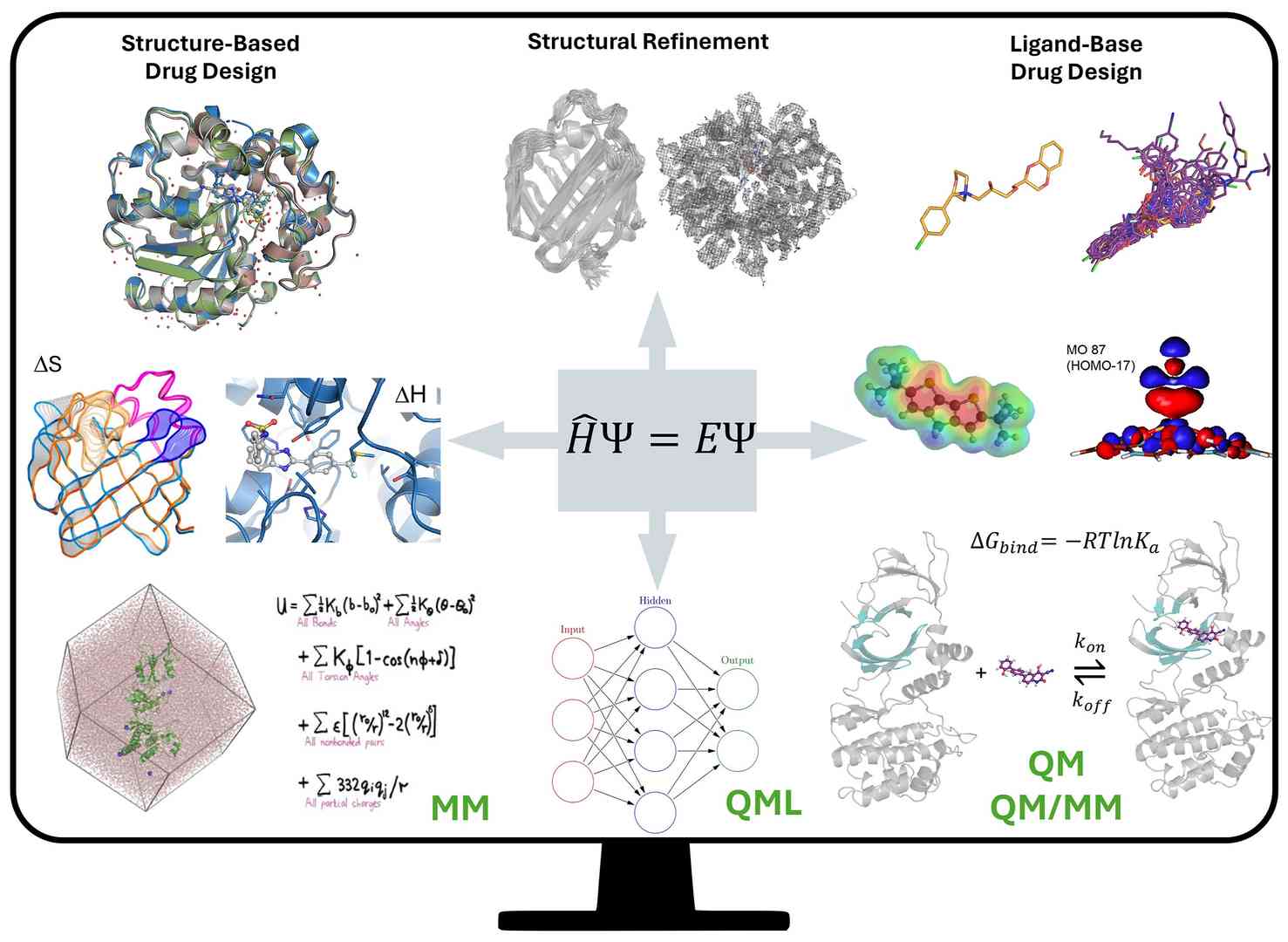 Figure 1. Schematic representation of the permeation of QM-based strategies in computer-aided drug design. (Ginex et al., 2024)
Figure 1. Schematic representation of the permeation of QM-based strategies in computer-aided drug design. (Ginex et al., 2024)
Conventional molecular mechanics (MM) and empirical scoring functions treat molecules as ball-and-spring models, with forces calculated from potential energy functions that depend on bond lengths, bond angles, torsions, and pairwise non-bonded terms. While extremely fast, this approximation breaks down whenever the physics of binding depends on electrons: polarization, charge transfer, protonation states, metal-ligand coordination, π–π stacking, halogen bonding, and covalent inhibition. QM-based drug design addresses precisely these challenges.
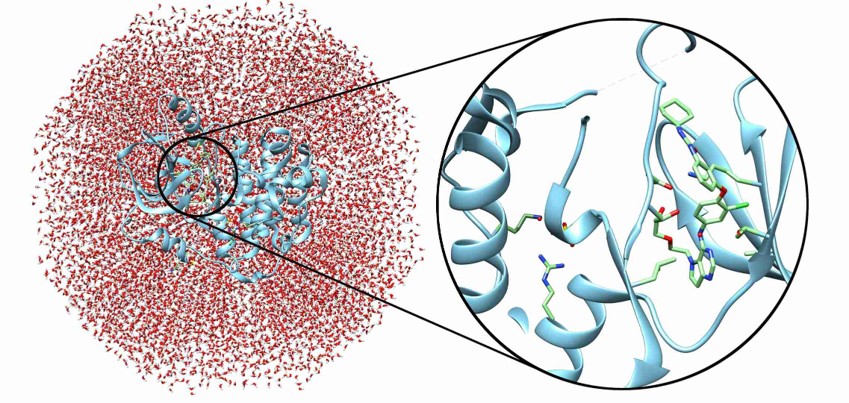 Figure 2. Protein–ligand complex in an explicit solvent environment. The enlarged view highlights the binding pocket used for quantum mechanics (QM) and QM/MM analyses to investigate molecular interactions and binding mechanisms.
Figure 2. Protein–ligand complex in an explicit solvent environment. The enlarged view highlights the binding pocket used for quantum mechanics (QM) and QM/MM analyses to investigate molecular interactions and binding mechanisms.
| Service Component | Description |
| Ab Initio Force Field Parameter Generation | Custom parameter derivation for non-standard residues, including RESP/CM5 charge fitting, torsion parameter optimization at the HF/6-31G* and B3LYP/6-311+G** levels, and derivation of bonded terms from Hessian calculations on small model compounds. |
| QM/MM Binding Calculations | Quantum treatment of the ligand and key binding-site residues combined with molecular mechanics treatment of the protein and solvent, enabling binding free energy estimation, per-residue energy decomposition, and characterization of polarization and charge-transfer effects. |
| Electronic Structure Analysis | Detailed analysis of charge distribution, frontier molecular orbitals (HOMO/LUMO), reactivity descriptors (electrophilicity, chemical potential, hardness), and Fukui functions to identify reactive sites and rationalize structure–activity relationships. |
| QM-Based Virtual Screening | GPU-accelerated quantum scoring of compound libraries, exploration of diverse chemical spaces, and reactivity-based filtering to prioritize candidates whose binding is governed by electronic effects rather than shape complementarity alone. |
| Ligand Optimization & Mechanism Elucidation | Transition-state modeling, reaction pathway analysis, and covalent binding mechanism studies, including activation energy and reaction-rate prediction for covalent inhibitors and prodrug activation workflows. |
Background:
A client was optimizing a series of inhibitors targeting a zinc metalloprotease. Standard empirical force fields failed to reproduce the coordination geometry around the catalytic Zn2+ ion and produced binding-energy rankings inconsistent with experimental SAR, stalling lead optimization.
Our Solution:
Profacgen constructed a QM/MM system in which the zinc ion, the zinc-binding group of the ligand, and the coordinating histidine residues were placed in the QM region (B3LYP/6-31G*), while the remainder of the protein and solvent were treated with molecular mechanics. Charge-fitting and cation-specific Lennard-Jones parameters were derived ab initio for the zinc environment, and binding free energies were computed using QM/MM-PBSA with per-residue energy decomposition.
Final Results:
The QM/MM model reproduced the key coordination geometry and predicted binding energies within 1 kcal/mol of the experimental values. Decomposition analysis pinpointed the dominant contribution of the zinc-binding group and guided substitution of the hydroxamate warhead, leading to a 6-fold improvement in potency against the primary target while preserving selectivity over related metalloproteases.
Background:
A client developing a kinase covalent inhibitor targeting a non-conserved cysteine residue observed potent cellular activity but suffered from off-target reactivity that limited the therapeutic window. Classical docking could not explain the selectivity profile or rank warhead modifications.
Our Solution:
Profacgen modeled the Michael addition reaction pathway between the acrylamide warhead and the cysteine thiol using DFT (ωB97X-D/6-311+G**) for the reacting moiety embedded in a QM/MM representation of the kinase active site. Activation energies, reaction rates, and intrinsic electrophilicities were computed for the parent scaffold and a panel of warhead analogs, and Fukui functions were used to map the local reactivity of each candidate.
Final Results:
QM calculations accurately predicted the activation energy and reproduced the experimental reaction rate within a factor of two. Based on the reactivity descriptors, Profacgen identified warhead substitutions that reduced intrinsic electrophilicity while preserving productive orientation in the target binding pocket. The redesigned inhibitor improved selectivity for the intended cysteine by 10-fold and restored a viable therapeutic window.
References:
Fill out this form and one of our experts will respond to you within one business day.