We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our
Privacy Policy
Profacgen offers ATTEC development services, providing autophagosome-tethering compounds for selective degradation of intracellular proteins, protein aggregates, and organelles through direct LC3-mediated autophagy, independent of the ubiquitin-proteasome system.
Conventional PROTACs and molecular glues rely on E3 ubiquitin ligase recruitment and proteasomal degradation, limiting their applicability to proteins with accessible ubiquitination sites and adequate proteasome engagement. Protein aggregates, misfolded proteins, and large macromolecular assemblies often evade proteasomal recognition and accumulate in neurodegenerative and proteinopathy disorders.
ATTEC (autophagosome-tethering compound) technology offers a fundamentally distinct mechanism: small-molecule bridges that simultaneously bind a target protein and the autophagosomal membrane protein LC3, tethering cargo directly to forming autophagosomes for lysosomal degradation. This ubiquitin-independent approach enables clearance of aggregates and soluble proteins alike, with particular promise for neurodegenerative disease targets.
What Is ATTEC?
ATTEC is a novel targeted protein degradation strategy based on the macroautophagy pathway. The platform comprises three defining features:
Autophagy-targeting chimeras: ATTECs are small-molecule heterobifunctional compounds designed to interact with both a target protein and LC3, a core autophagy protein. Unlike PROTACs, which require ubiquitination, ATTECs directly tether targets to autophagosomes without engaging the ubiquitin-proteasome system
Selective autophagy: ATTECs harness the autophagy machinery with high selectivity. By bridging specific target proteins to LC3, they recruit cargo to forming phagophores without perturbing global autophagy flux or inducing non-specific bulk degradation. This precision minimizes off-target effects on cellular homeostasis
Intracellular degradation: ATTECs operate within the cytoplasm and nucleus, targeting soluble proteins, protein aggregates, and organelles for autophagic degradation. The LC3-tethering mechanism is compatible with diverse target classes, including those resistant to proteasomal processing
Mechanism of ATTEC-Mediated Degradation
ATTEC-mediated degradation proceeds through direct autophagosomal recruitment without ubiquitin involvement:
Target Recognition: The target-binding moiety of the ATTEC engages the protein of interest—whether a soluble cytosolic protein, nuclear factor, or aggregated misfolded species—with high affinity and specificity.
Autophagy Tagging via LC3 Engagement: The LC3 ligand moiety of the ATTEC binds to LC3-I or LC3-II, the lipidated form conjugated to phosphatidylethanolamine (PE) and anchored on autophagosomal membranes. This dual engagement forms a ternary complex: target protein–ATTEC–LC3.
Autophagosome Formation and Sequestration: The tethered target protein is engulfed by the expanding phagophore membrane. LC3-II on the inner membrane retains the cargo as the autophagosome matures and seals, isolating the target from the cytoplasm.
Lysosomal Degradation: The autophagosome fuses with a lysosome, forming an autolysosome. Lysosomal hydrolases degrade the sequestered target protein into amino acids. The ATTEC molecule is released and can catalyze additional degradation cycles.
Figure 1. The concept of ATTECs. ATTEC molecules bridge target proteins to LC3 on autophagosomal membranes, inducing selective autophagic degradation without ubiquitin involvement. (Adapted from Ding et al., 2020)
Our ATTEC Development Services
Profacgen provides comprehensive ATTEC development from molecular design to mechanistic validation:
ATTEC Design
Rational design of heterobifunctional molecules optimized for target binding and LC3 engagement.
Target warhead selection: small-molecule ligands, fragment hits, or peptide binders with validated target engagement
LC3 ligand optimization: structure-based design of LC3-binding moieties with high affinity and selectivity over GABARAP and GATE-16 family proteins
Linker engineering: optimization of length, flexibility, and chemical composition to maximize ternary complex stability and cellular permeability
Ligand Development
Synthesis and analytical characterization of ATTEC components and full conjugates.
LC3 ligand synthesis and validation by SPR, ITC, and fluorescence polarization
Target warhead scale-up and purity confirmation by HPLC and LC-MS
Full ATTEC assembly via click chemistry, amide coupling, or bioorthogonal conjugation strategies
Activity Evaluation
Cellular assessment of target degradation efficiency and selectivity.
Western blot and quantitative mass spectrometry for target protein level quantification
DC50 and Dmax determination across dose-response and time-course experiments
Autophagy flux monitoring: LC3-II turnover, p62/SQSTM1 degradation, and autophagosome accumulation by flow cytometry and imaging
Mechanism Validation
Confirmation of autophagy-dependent, ubiquitin-independent degradation.
Autophagy inhibition rescue: chloroquine, bafilomycin A1, and ATG5/ATG7 knockout to confirm autophagy dependence
Proteasome independence: MG132 and bortezomib treatment to rule out UPS contribution
LC3 colocalization: confocal microscopy with GFP-LC3 or mCherry-LC3 reporters to visualize target recruitment to autophagosomes
Applications
ATTEC technology addresses therapeutic and research challenges beyond the reach of proteasome-dependent modalities:
Protein Degradation: Elimination of soluble intracellular proteins, including transcription factors, kinases, and scaffold proteins that lack conventional druggable features or are resistant to proteasomal processing. ATTECs provide an orthogonal degradation route independent of ubiquitination
Organelle Clearance: Selective removal of damaged mitochondria (mitophagy), lipid droplets (lipophagy), and protein aggregates through LC3-mediated tethering. ATTECs can be engineered to target organelle-specific proteins, redirecting the autophagy machinery to discrete subcellular compartments
Disease Modeling: Neurodegenerative diseases—Alzheimer's, Parkinson's, and Huntington's disease—are characterized by accumulation of misfolded protein aggregates and oligomers. ATTECs have demonstrated significant reduction of mutant huntingtin protein aggregates, providing entry points to new therapeutic strategies. Molecules with high lipid solubility and molecular weight under 500 Da are particularly suited for blood-brain barrier penetration via passive diffusion
Advantages of ATTEC
Potentially Broad Target Spectrum: ATTECs can degrade proteins regardless of ubiquitination status, including aggregate-prone species, long-lived proteins, and macromolecular assemblies that evade proteasomal recognition.
Direct Targeting to Degradation Machinery: By tethering targets directly to LC3 on autophagosomal membranes, ATTECs bypass the requirement for ubiquitin ligase recruitment and polyubiquitin chain assembly.
Potentially Effective in All Cell Types: The autophagy pathway is conserved across eukaryotic cell types. ATTEC activity is not constrained by cell-type-specific E3 ligase expression profiles that limit PROTAC efficacy.
Low Molecular Weight: ATTECs are typically smaller than antibody-based degraders and comparable to small-molecule PROTACs, facilitating cellular permeability and oral bioavailability.
Ubiquitin-Independent Mechanism: Degradation proceeds without target ubiquitination, avoiding competition with deubiquitinases and enabling clearance of proteins with limited lysine accessibility.
Why Choose Profacgen
One-Stop Degradation Platform: Profacgen integrates ATTEC design, synthesis, and validation within a single workflow, eliminating vendor coordination overhead and accelerating project timelines.
LC3 Structural Biology Expertise: Our team leverages deep knowledge of LC3 family proteins (LC3A, LC3B, LC3C, GABARAP, GATE-16) to design selective ligands and avoid off-target autophagy protein engagement.
Aggregate Target Experience: Specialized capabilities for misfolded protein and inclusion body targeting, including huntingtin, tau, and α-synuclein aggregate degradation assessment.
Mechanistic Rigor: Comprehensive pathway validation including autophagy dependence, proteasome independence, and LC3 colocalization confirmation ensures unambiguous mechanism assignment.
BBB Penetration Support: For neurodegenerative applications, we provide physicochemical optimization (cLogP, PSA, molecular weight) and in silico blood-brain barrier permeability prediction.
Scenario 1: Mutant Huntingtin Aggregate Clearance by ATTEC
Program Context:
A Huntington's disease research program sought to reduce mutant huntingtin (mHTT) protein aggregates in neuronal models. Proteasome-based approaches were ineffective against insoluble aggregate species, and the team required a modality capable of engaging large macromolecular assemblies.
Objective:
To design an ATTEC that bridges mHTT aggregates to LC3, validate autophagy-dependent aggregate clearance in cellular models, and assess neuroprotective phenotypic outcomes.
Approach:
Profacgen identified a small-molecule mHTT binder through fragment screening and tethered it to an optimized LC3 ligand via a PEG-based linker. The ATTEC was validated for LC3 engagement by SPR (Kd = 180 nM) and for mHTT binding by fluorescence polarization. In mHTT-expressing neuronal cell lines, the ATTEC reduced soluble mHTT levels by >70% and aggregate burden by >60% at 48 hours, as quantified by filter retardation assay and immunofluorescence.
Outcome:
Degradation was completely blocked by bafilomycin A1 (autophagy inhibitor) but unaffected by MG132 (proteasome inhibitor), confirming autophagy specificity. GFP-LC3 colocalization with mHTT aggregates was observed by live-cell imaging. Neuronal viability improved significantly, supporting ATTEC-mediated aggregate clearance as a viable therapeutic strategy for Huntington's disease.
Scenario 2: Soluble Kinase Degradation via LC3 Tethering
Program Context:
An oncology program required degradation of a cytosolic kinase that lacked suitable E3 ligase recruiters for PROTAC development. The kinase was poorly ubiquitinated under basal conditions, and alternative degradation modalities were sought.
Objective:
To develop an ATTEC targeting the kinase via its ATP-binding site, demonstrate LC3-mediated autophagic degradation, and assess pathway engagement and phenotypic consequences.
Approach:
Profacgen repurposed a known kinase inhibitor as the target warhead and conjugated it to a selective LC3B ligand identified by fragment-based screening. The ATTEC was validated for dual binding by SPR and crystallography. Cellular degradation was assessed by Western blot and quantitative proteomics across multiple cancer cell lines.
Outcome:
The ATTEC achieved >80% kinase degradation at 500 nM with DC50 of 120 nM. Degradation was blocked by ATG7 knockout and chloroquine, confirming autophagy dependence. Downstream signaling (p-ERK, p-S6) was suppressed, and cell proliferation was inhibited. The study established ATTEC as a viable alternative to PROTACs for targets with limited ubiquitination.
Q: What is the difference between ATTEC and PROTAC?
A: PROTACs recruit E3 ubiquitin ligases to induce proteasomal degradation and require target ubiquitination. ATTECs directly tether target proteins to LC3 on autophagosomal membranes, bypassing ubiquitin entirely. This enables ATTEC to degrade proteins that are poorly ubiquitinated, form aggregates, or exceed proteasomal size limits. PROTACs are generally preferred for soluble intracellular proteins with accessible lysines; ATTECs excel for aggregates, organelles, and ubiquitin-resistant targets.
Q: What is the difference between ATTEC and AUTAC?
A: AUTACs introduce K63-linked ubiquitin tags to recruit autophagy receptors (p62/SQSTM1, NDP52) and induce selective autophagy. ATTECs directly bind LC3 on autophagosomal membranes without ubiquitin involvement. AUTACs modify the target with a ubiquitin mimic; ATTECs act as molecular bridges. ATTECs are generally smaller and mechanistically simpler, while AUTACs may offer additional signaling specificity through receptor recruitment.
Q: What is LC3 and why is it central to ATTEC mechanism?
A: LC3 (microtubule-associated protein 1A/1B-light chain 3) is a core autophagy protein. Cytosolic LC3-I is conjugated to phosphatidylethanolamine (PE) to form LC3-II, which is recruited to autophagosomal membranes. LC3-II mediates membrane expansion, cargo recognition, and autophagosome closure. ATTECs bind LC3-II (or LC3-I) to tether target proteins directly to forming autophagosomes, bypassing the need for ubiquitin tags or autophagy receptors.
Q: What types of proteins can ATTEC degrade?
A: ATTECs can degrade soluble cytosolic proteins, nuclear proteins, protein aggregates, and organelle-associated proteins. They are particularly effective for misfolded protein aggregates (huntingtin, tau, α-synuclein), large multi-protein complexes, and proteins resistant to proteasomal degradation. ATTECs are not suitable for extracellular or membrane proteins, which are better addressed by LYTACs.
Q: What sample requirements are needed for ATTEC development?
A: For design, a validated target ligand or structural information is required. For synthesis, no biological sample is needed. For validation, target-expressing cell lines are essential. Profacgen can assist with construct generation, stable cell line development, and aggregate-prone model systems. For mechanism validation, access to autophagy inhibitors (chloroquine, bafilomycin A1) and LC3 reporter systems is required.
Q: Can ATTEC cross the blood-brain barrier for neurodegenerative applications?
A: ATTECs are small molecules with potential for blood-brain barrier (BBB) penetration. Profacgen optimizes ATTEC physicochemical properties (molecular weight <500 Da, high lipophilicity, low polar surface area) to favor passive diffusion across the BBB. For Huntington's disease and related proteinopathies, ATTECs have demonstrated aggregate reduction in cellular models. In vivo BBB penetration requires further pharmacokinetic optimization and is evaluated on a project-specific basis.
References:
Ding Y, Fei Y, Lu B. Emerging new concepts of degrader technologies. Trends in Pharmacological Sciences. 2020;41(7):464-474. doi:10.1016/j.tips.2020.04.005
Online Inquiry
Fill out this form and one of our experts will respond to you within one business day.
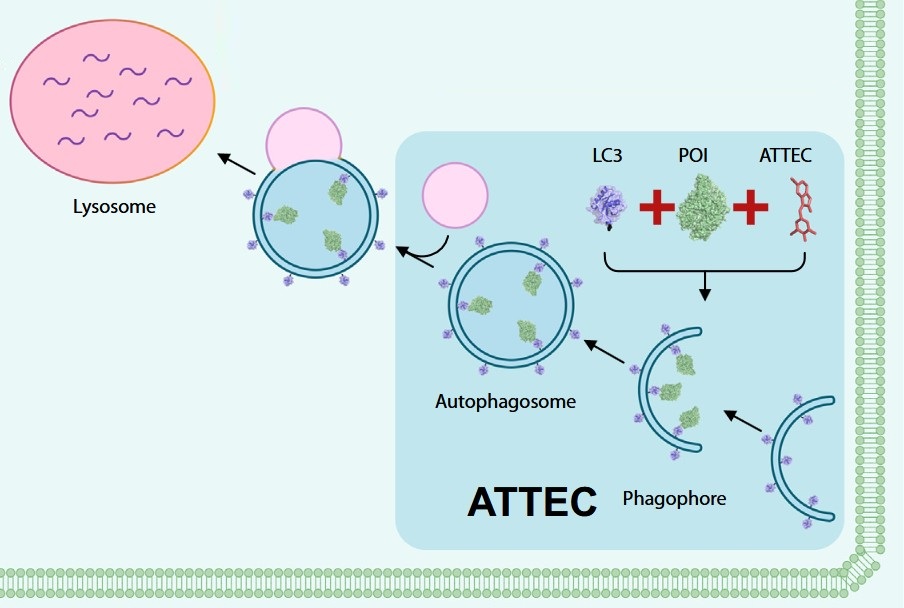 Figure 1. The concept of ATTECs. ATTEC molecules bridge target proteins to LC3 on autophagosomal membranes, inducing selective autophagic degradation without ubiquitin involvement. (Adapted from Ding et al., 2020)
Figure 1. The concept of ATTECs. ATTEC molecules bridge target proteins to LC3 on autophagosomal membranes, inducing selective autophagic degradation without ubiquitin involvement. (Adapted from Ding et al., 2020)