The interactions between proteins and peptides play essential functional roles in living organisms and they can be found in a variety of signaling pathways involved in cellular localization, immune response or protein expression and degradation. Erroneous protein-peptide interactions are associated with a number of diseases (e.g., cancer, autoimmune diseases). The understanding of protein-peptide interactions is critical for targeted drug design and their structural characterization is a hot subject of current experimental and theoretical research.
Profacgen makes use of state-of-the-art docking software tools to model protein–peptide interactions and to predict the complex structure. The general modeling procedure is divided into two stages: (1) a global search is performed to predict the binding site at the protein receptor surface; (2) a flexible local docking method is used to fit the peptide structure into the known binding site. These two steps take into consideration the significant conformational changes upon binding of the two components. The peptide folding process is explicitly simulated in the docking protocol. When experimental or predicted data on binding site residues are available, such information can be used to constrain the docking to local regions of the protein surface. Finally, the best-scored structures are clustered, ranked and representatives of the largest clusters are selected for high resolution structural refinement before being presented to the customer.
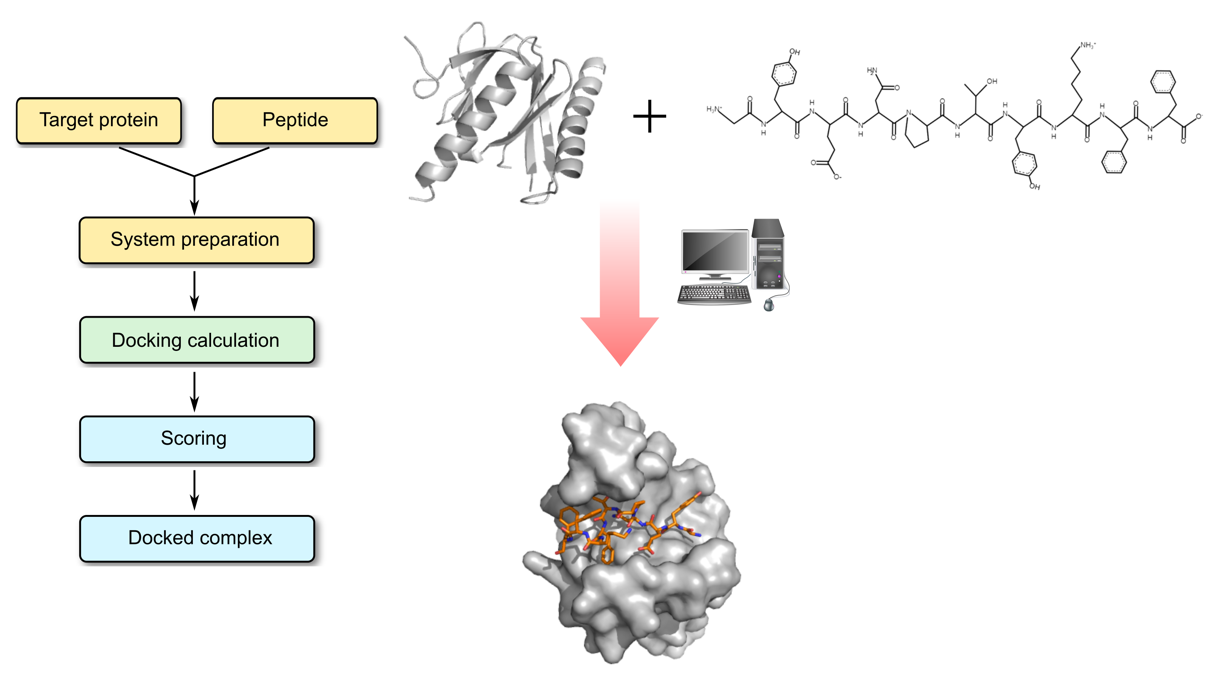
Profacgen employs computational docking techniques to search all possible binding modes in the translational and rotational space between a protein receptor and its peptide binding partner. Given a protein receptor structure and a peptide sequence, we predict the structure of protein–peptide complex, starting from random conformations and positions of the peptide. This is an important means for understanding the physicochemical forces that underlie macromolecular interactions and a valuable tool for modeling protein–peptide complex structures at the atomic level. Furthermore, the precise understanding of these interactions will facilitate the rational design of potentially therapeutic peptide.
We provide the service in a customizable fashion to suit our customers’ specific research goals. Please do not hesitate to contact us for more details about our protein–peptide docking service.
Fill out this form and one of our experts will respond to you within one business day.