We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our
Privacy Policy
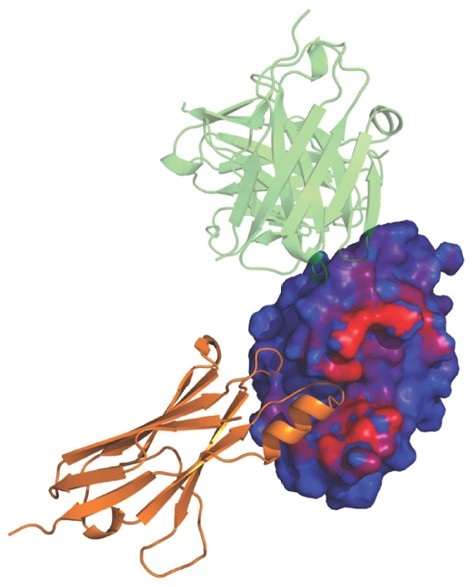
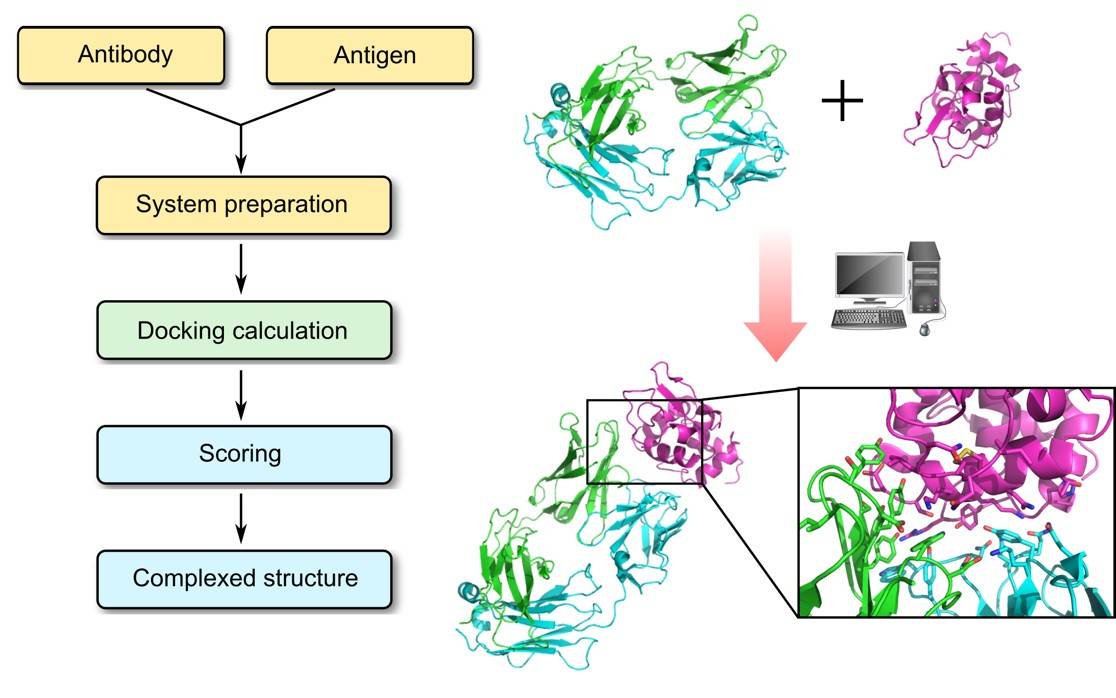
Antibodies are essential recognition elements of the immune system, binding tightly and specifically to their target antigens to block pathogenic activity or mark foreign entities for destruction by effector cells. In recent decades, antibodies have increasingly been harnessed as powerful diagnostic tools and therapeutic drugs across a wide range of diseases, including cancer, autoimmune disorders, and infectious diseases. The ability to engineer antibodies with improved affinity, specificity, and stability hinges critically on understanding the structural basis of antibody–antigen recognition. At Profacgen, our Antibody–Antigen Docking service provides a fast, cost-effective computational pipeline for generating high-quality three-dimensional models of antibody–antigen complexes, enabling researchers to gain mechanistic insights and rationally guide antibody engineering efforts.
While X-ray crystallography remains the gold standard for antibody–antigen complex structures, co-crystallization is often hampered by poor crystal quality, interfacial conformational heterogeneity, and high resource demands. Computational docking offers a rapid, cost-effective alternative. Our protocol prioritizes CDR loop flexibility—especially the H3 loop, which dominates binding specificity and affinity. Through a pipeline of rigid-body global search, constraint integration, antibody-specific scoring, clustering, and energy minimization, Profacgen delivers reliable structural models to guide experimental follow-up.
Why Antibody–Antigen Docking?
Understanding the atomic-level details of antibody–antigen interactions is fundamental to both basic immunological research and applied antibody engineering. The physicochemical forces that govern macromolecular recognition—including hydrogen bonding, electrostatic complementarity, van der Waals interactions, and hydrophobic packing—can only be fully appreciated through three-dimensional structural models. Computational antibody–antigen docking provides a systematic framework for exploring these interactions, enabling researchers to:
Identify key contact residues and binding hot spots at the antibody–antigen interface
Map conformational and linear epitopes recognized by a given antibody
Rationalize mutational data and structure–activity relationships
Guide affinity maturation and specificity engineering campaigns
Assess the structural consequences of antibody humanization or CDR grafting
Generate testable hypotheses for experimental validation, including alanine scanning mutagenesis
Accelerate lead candidate selection in early-stage antibody discovery programs
By integrating computational docking with experimental data, researchers can significantly reduce the trial-and-error burden inherent in antibody optimization and focus resources on the most promising engineered variants.
Our Antibody–Antigen Docking Service Offerings
Services
Details
Antibody Modeling & CDR Refinement
Fv, Fab, and full-length antibody structural modeling from sequence
CDR loop grafting onto framework templates with iterative refinement
Support for multiple antibody numbering schemes: Chothia, Kabat, IMGT, and Aho
Specialized H3 loop conformational sampling and energy minimization
Antigen Structure Preparation
Curation and preparation of experimentally determined antigen structures
Homology modeling of antigen when experimental structure is unavailable
Epitope region definition and binding site mapping based on available data
Addition of missing loops, side chains, and co-factors as needed
Antibody–Antigen Docking
Rigid-body global conformational search of antibody–antigen docking poses
Antibody-specific scoring function dedicated to antigen–antibody interactions
Integration of user-defined restraints (experimental epitope data, mutagenesis results) to limit search space
Solvated docking protocol with explicit treatment of interfacial water molecules
Clustering, Epitope Mapping & Selection
Energy-based clustering to identify highly populated low-energy conformational basins
Computational epitope mapping identifying residues contributing to binding
Paratope analysis detailing CDR residue contributions to antigen recognition
Filtering of output predictions based on user-defined structural or biochemical restraints
Refinement & Quality Assessment
Energy minimization of representative models from top-ranked clusters
Model quality assessment by multiple criteria (interface energy, shape complementarity, residue-level scoring)
Interaction interface characterization—hydrogen bond networks, salt bridges, hydrophobic contacts
Support for modified amino acids and non-standard residues in both antibody and antigen
Antibody-Specific Scoring Function: Our docking pipeline employs a scoring function specifically optimized for assessing antibody–antigen interactions, incorporating knowledge-based potentials derived from experimentally determined complex structures. This ensures that the energetically most favorable models reflect biologically relevant binding modes rather than generic protein–protein docking solutions.
Explicit Solvent Treatment: The solvated docking protocol explicitly models interfacial water molecules, which often mediate critical hydrogen-bonding networks at antibody–antigen interfaces. By accounting for the thermodynamic contribution of ordered water, our approach captures binding determinants that are missed by implicit solvent methods.
CDR Loop Flexibility & Refinement: We place special emphasis on the conformational flexibility of the CDR loops, particularly the H3 loop, which is the most variable in sequence, length, and structure. Our protocol includes dedicated loop modeling and refinement steps to explore the conformational landscape of the antigen-binding site before and during docking.
User-Guided Restraint Integration: Docking predictions can be filtered and ranked based on experimentally derived restraints—such as epitope-mapped residues from mutagenesis, competition assays, or hydrogen-deuterium exchange data. This hybrid computational-experimental approach dramatically improves the accuracy and reliability of predicted complex models.
Comprehensive Numbering Scheme Support: Our platform is fully compatible with the major antibody numbering schemes—Chothia, Kabat, IMGT, and Aho—ensuring seamless integration with existing antibody engineering workflows and facilitating communication of results across different research teams and database annotations.
Case 1: Epitope Mapping of a Neutralizing Antibody Against a Viral Glycoprotein
Background:
A client had isolated a potent neutralizing monoclonal antibody targeting the surface glycoprotein of an emerging virus. While functional assays confirmed potent neutralization activity, the molecular basis of this activity—including which specific residues constituted the epitope—remained unknown, hindering structure-based vaccine design efforts.
Solution:
Using our antibody–antigen docking pipeline, we generated over 10,000 docked conformations of the antibody Fv region against the trimeric glycoprotein ectodomain. The docking was guided by prior knowledge that the antibody competed with the host receptor for binding, narrowing the search to the receptor-binding domain. Clustering of top-scoring models revealed a dominant binding pose, and computational epitope mapping identified a conformational epitope spanning three discontinuous loops on the glycoprotein surface. Paratope analysis showed that the heavy chain CDR H3 loop contributed the majority of binding energy, with supplementary contacts from H1, H2, and the light chain CDR L3 loop.
Results:
The predicted epitope residues were validated through alanine-scanning mutagenesis, with 11 of 14 predicted contact residues showing significant reduction in antibody binding upon mutation. The structural model directly guided the design of a stabilized immunogen focusing the immune response on the identified neutralizing epitope, accelerating the client's vaccine development program.
Case 2: Structural Assessment of CDR Grafting for Therapeutic Antibody Humanization
Background:
A client sought to humanize a murine monoclonal antibody with promising anti-tumor activity for clinical development. Standard CDR grafting onto a human germline framework had resulted in a significant loss of binding affinity, and the structural basis for this loss was unclear. The client needed to identify which framework residues required back-mutation to the original murine sequence to restore affinity while minimizing the risk of immunogenicity.
Solution:
We built structural models of the chimeric antibody (murine variable domains on human constant domains), the initial CDR-grafted variant, and the fully murine antibody. Antibody–antigen docking was performed for each variant against the target antigen. Comparative analysis of the docking models revealed that two framework residues in the heavy chain (positions H71 and H78, Kabat numbering) underwent subtle conformational changes upon grafting, which propagated to alter the orientation of the CDR H2 loop and disrupted a critical salt bridge with the antigen. Additional analysis identified a Vernier zone residue (H49) whose replacement affected the packing of CDR H2 against the framework.
Results:
Based on our structural analysis, the client performed targeted back-mutations at positions H49, H71, and H78 in the humanized construct. The triple back-mutant recovered full binding affinity comparable to the parental murine antibody, while maintaining a high degree of humanness (assessed by in silico T-cell epitope prediction). The humanized candidate progressed to lead optimization with a significantly reduced immunogenicity risk profile.
Q: What input data is required to perform antibody–antigen docking?
A: Minimum input: amino acid sequences of the antibody variable domains (heavy and light chains) and either the structure or sequence of the target antigen. Experimental epitope data (mutagenesis, competition assays, HDX-MS) can be incorporated as restraints to improve accuracy. Additional details on numbering scheme, CDR definitions, or PTMs are also valuable.
Q: How reliable are computationally predicted antibody–antigen complex structures?
A: Reliability depends on input quality, binding-induced conformational changes, and available restraints. With appropriate restraints, benchmark studies show near-native predictions (interface RMSD < 2.0 Å) for many targets. We assess each model via interface energy, shape complementarity, and contact analysis, and report confidence levels transparently. Models are best viewed as testable hypotheses to guide experiments.
Q: How do you handle the conformational flexibility of CDR loops, especially the H3 loop?
A: H3 is the most variable loop. Our approach: generate an H3 ensemble using high-resolution loop modeling; treat selected conformations as discrete alternatives in multi-conformation docking; and refine top models via side-chain repacking and energy minimization. Other CDR loops are sampled from canonical conformations based on structural databases.
Q: What antibody numbering schemes do you support, and why does it matter?
A: We support Chothia, Kabat, IMGT, and Aho. Each defines CDR boundaries differently, affecting binding-site annotation. Kabat is sequence-based and widely used; Chothia is structure-based; IMGT is unified for immunoglobulins and TCRs; Aho optimizes both sequence and structure alignment. We accept any scheme and deliver results in your preferred format.
Q: Can you dock antibodies to antigens for which no experimental structure is available?
A: Yes. If no antigen structure is available, we can generate a homology model provided a suitable template exists. Accuracy depends on template quality. For antigens without templates, we may recommend ab initio prediction for small domains or focus on structured subdomains. We always communicate expected confidence levels to inform downstream validation.
Q: What deliverables do I receive, and in what format?
A: Standard delivery includes: (1) top complex models in PDB format; (2) a detailed protocol and quality assessment report; (3) epitope–paratope contact maps; (4) publication-quality figures; and (5) PyMOL session files. Custom deliverables (e.g., docking trajectories, energy decomposition, multi-variant comparisons) can be arranged upon request.
References:
Desta IT, Kotelnikov S, Jones G, et al. The ClusPro AbEMap web server for the prediction of antibody epitopes. Nat Protoc. 2023;18(6):1814-1840. doi:10.1038/s41596-023-00826-7
Online Inquiry
Fill out this form and one of our experts will respond to you within one business day.