We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our
Privacy Policy
The tertiary and quaternary structures of a protein define its functional architecture—governing active-site geometry, subunit assembly, and interactions with ligands, receptors, and effector molecules. Profacgen's Tertiary & Quaternary Structure Characterization services integrate orthogonal biophysical and mass spectrometry platforms to deliver comprehensive higher-order structure (HOS) verification. Whether confirming the folded conformation of a monoclonal antibody, monitoring oligomeric state during formulation development, or demonstrating structural similarity in a biosimilar comparability exercise, our multi-method approach provides the resolution, sensitivity, and regulatory documentation required to advance biopharmaceutical programs from early development through commercial release.
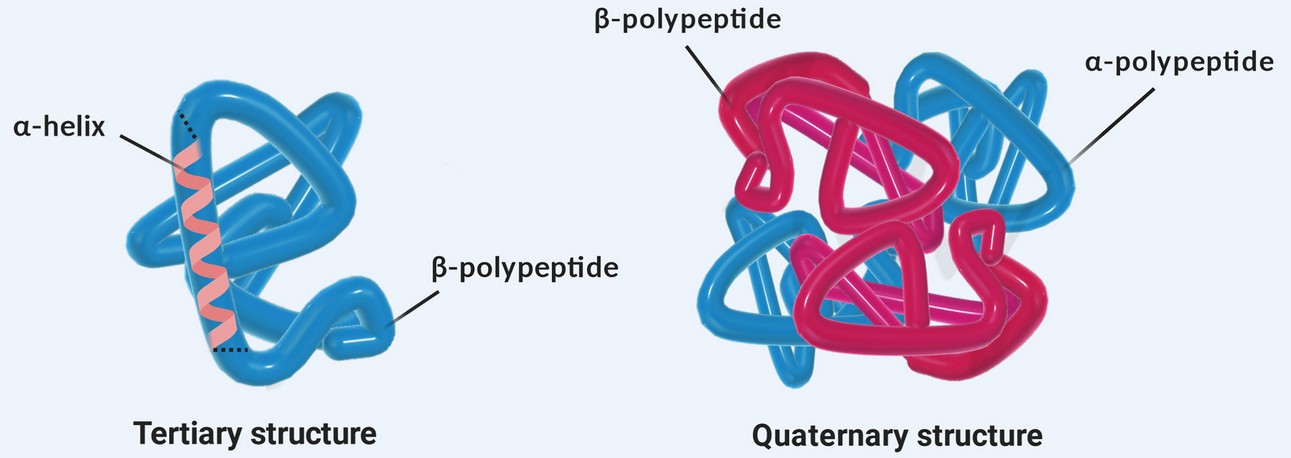
Biopharmaceutical efficacy and safety are intrinsically linked to correct protein folding and assembly. The tertiary structure encompasses the three-dimensional arrangement of a single polypeptide chain, including the positioning of aromatic residues, disulfide-bonded domains, and ligand-binding pockets. The quaternary structure describes the association of multiple subunits—such as the heterotetrameric assembly of two heavy and two light chains in an IgG antibody or the reversible oligomerization of a cytokine receptor. Regulatory agencies classify HOS as a critical quality attribute (CQA) and mandate its characterization for all recombinant protein therapeutics and biosimilars.
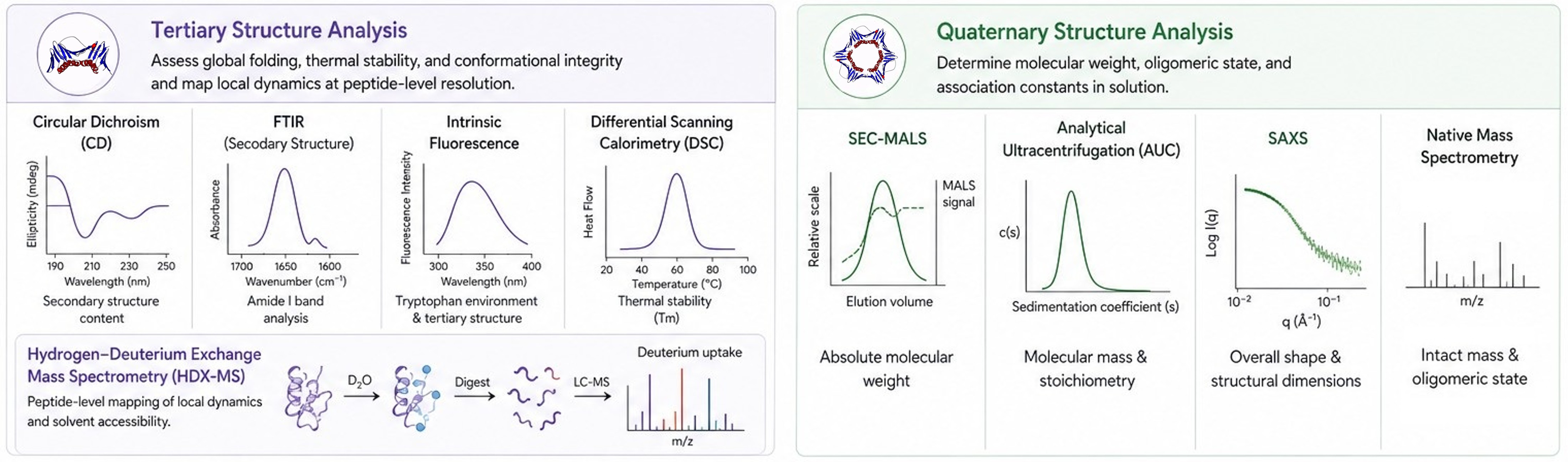
Profacgen addresses these requirements through a complementary multi-platform strategy. Tertiary structure is interrogated by circular dichroism (CD), Fourier-transform infrared spectroscopy (FTIR), intrinsic fluorescence, and differential scanning calorimetry (DSC) to assess global folding, thermal stability, and conformational integrity. For site-specific resolution, hydrogen–deuterium exchange mass spectrometry (HDX-MS) maps local dynamics and solvent accessibility at peptide-level resolution. Quaternary structure is evaluated by size-exclusion chromatography with multi-angle light scattering (SEC-MALS), analytical ultracentrifugation (AUC), small-angle X-ray scattering (SAXS), and native mass spectrometry to determine molecular weight, oligomeric state, and association constants in solution.
Our Tertiary & Quaternary Structure Characterization Service Offerings
Profacgen provides comprehensive HOS characterization solutions tailored to research, development, and quality control applications. Our offerings include:
Service Component
Description
Tertiary Structure by Circular Dichroism (CD)
Far-UV CD (190–260 nm) for secondary structure quantification (α-helix, β-sheet, turns, random coil)
Near-UV CD (250–320 nm) for tertiary structure assessment via aromatic residue environments
Temperature-ramp CD for thermal unfolding transitions and apparent melting temperatures (Tm)
Buffer and pH screening to identify conditions that preserve native conformation
Tertiary Structure by FTIR & Fluorescence Spectroscopy
FTIR amide I band deconvolution (1,600–1,700 cm−1) for secondary structure composition in solid and solution states
Intrinsic tryptophan fluorescence emission scans to probe local tertiary environment and conformational changes
Extrinsic fluorescence (ANS, Nile Red) for detection of partially unfolded or aggregation-prone species
Lyophilized formulation analysis to verify cake structure stability and moisture impact on HOS
Thermal Stability by Differential Scanning Calorimetry (DSC)
High-sensitivity DSC for direct measurement of thermodynamic unfolding parameters (Tm, ΔH, ΔCp)
Multi-domain resolution of CH2, CH3, and Fab unfolding transitions in monoclonal antibodies
Formulation screening to rank excipients by conformational stabilization potency
Forced degradation correlation: DSC Tm shifts as an early indicator of structural perturbation
Site-Specific HOS by Hydrogen–Deuterium Exchange MS (HDX-MS)
Bottom-up HDX-MS with automated LC-MS/MS for peptide-level deuterium uptake kinetics
Comparability and epitope mapping by differential HDX-MS (reference vs. test sample)
PLIMSTEX and SUPREX methodologies for ligand-binding affinity and thermodynamic stability
Data analysis with Deuteros or HDX Workbench for automated uptake curve generation and statistical testing
Quaternary Structure & Oligomeric State Analysis
SEC-MALS for absolute molar mass determination of monomers, dimers, and high-order oligomers
Analytical ultracentrifugation (AUC) by sedimentation velocity and equilibrium for shape and association constants
Small-angle X-ray scattering (SAXS) for low-resolution three-dimensional envelope and radius of gyration (Rg)
Native mass spectrometry (native MS) for direct observation of intact oligomers and subunit stoichiometry
Orthogonal method integration: Spectroscopic, thermodynamic, and hydrodynamic techniques provide convergent evidence of structural integrity
Site-specific HOS resolution: HDX-MS delivers residue-level conformational information unattainable by low-resolution biophysical methods alone
Regulatory compliance: Full alignment with ICH Q6B, FDA CMC guidance, and EMA biosimilar requirements for HOS demonstration
Comparability and biosimilarity support: Statistical similarity assessment for process changes, formulation transfers, and biosimilar analytical packages
Formulation and stability guidance: Thermal and colloidal stability data inform excipient selection, storage conditions, and container-closure compatibility
Representative Case Studies
Case 1: Biosimilar HOS Comparability for an Antibody Candidate
Background:
A biosimilar developer needed to demonstrate analytical similarity between their antibody candidate and the reference product as part of a totality-of-evidence submission. Higher-order structure was identified as a high-risk attribute because even subtle conformational differences could affect TNF-α binding, Fc receptor interactions, and long-term stability. The client required orthogonal HOS data spanning global, thermal, and site-specific readouts.
Our Solution:
Profacgen designed a comprehensive HOS comparability package. Far-UV and near-UV CD spectra were collected in triplicate and converted to mean residue ellipticity for secondary and tertiary structure overlay. FTIR amide I band deconvolution provided independent secondary structure estimates. DSC was performed to compare CH2, CH3, and Fab domain thermal transitions. HDX-MS was conducted at 0, 10, 60, and 240 minutes to generate differential uptake plots across the entire sequence. SEC-MALS confirmed identical monomeric mass and absence of soluble aggregates.
Final Results:
CD spectra were statistically superimposable with <2 % difference in β-sheet content. DSC Tm values for all three domains matched within 0.3 °C. HDX-MS differential analysis revealed no statistically significant uptake differences (>99 % confidence) at any peptide across the heavy or light chains. SEC-MALS confirmed a monomer mass of 148.1 kDa for both products with <0.5 % aggregate.
Case 2: Oligomeric State Verification of a Self-Associating Cytokine
Background:
A protein engineering company designed a mutant interleukin designed to form reversible dimers at high concentration for subcutaneous delivery. The intended mechanism relied on concentration-dependent self-association: monomer at low concentration for systemic distribution, dimer at high concentration for depot formation. The client needed to rigorously characterize the oligomeric equilibrium and confirm that the dimer interface matched the computational design.
Our Solution:
Profacgen deployed a multi-method quaternary structure package. SEC-MALS at 1, 5, 10, and 25 mg/mL revealed a concentration-dependent shift from monomer (Mw = 17.2 kDa) to dimer (Mw = 34.4 kDa), with an apparent dissociation constant (KD) of 4.8 µM. Sedimentation velocity AUC resolved two species with sedimentation coefficients of 1.8 S (monomer) and 2.6 S (dimer), confirming the stoichiometry. SAXS generated low-resolution envelopes showing excellent agreement with the computationally predicted dimer structure (χ2 = 1.12). Native MS under nondenaturing conditions directly observed both monomer and dimer charge-state envelopes.
Final Results:
All four orthogonal methods converged on a reversible, noncovalent dimer with 2:1 stoichiometry and a KD in the low-micromolar range—ideal for the proposed depot mechanism. SAXS envelope fitting confirmed that the dimer interface involved the engineered hydrophobic patch as designed. The client used Profacgen's data to support their IND submission and to establish release specifications for monomer/dimer ratio as a function of concentration. The project advanced to Phase I dosing within nine months.
Q: What is the difference between tertiary and quaternary structure?
A: Tertiary structure refers to the three-dimensional folding of a single polypeptide chain, encompassing the spatial arrangement of secondary structure elements, aromatic residues, disulfide bonds, and active-site loops. It is stabilized by hydrophobic interactions, hydrogen bonds, ionic interactions, and disulfide bridges. Quaternary structure describes the association of two or more polypeptide chains (subunits) into a functional complex—such as the heterotetrameric assembly of an IgG antibody or the homodimerization of a cytokine. While tertiary structure defines the fold of individual chains, quaternary structure governs inter-subunit contacts, stoichiometry, and oligomeric equilibrium.
Q: Which methods do you use for tertiary structure analysis, and how do they differ?
A: We employ an orthogonal panel. Circular dichroism (CD) in the far-UV region quantifies secondary structure composition (α-helix, β-sheet), while near-UV CD probes tertiary structure through aromatic residue environments. FTIR provides complementary secondary structure data, particularly for lyophilized formulations and samples in complex matrices. Intrinsic fluorescence monitors tryptophan exposure as a sensitive indicator of local tertiary changes. DSC measures thermal stability by detecting domain-specific unfolding transitions. HDX-MS offers the highest resolution, mapping solvent accessibility and conformational dynamics at the peptide level. Together, these methods provide convergent evidence of correct folding.
Q: How is quaternary structure analyzed, and what information does it provide?
A: Quaternary structure is analyzed by hydrodynamic and scattering methods that preserve native protein interactions. SEC-MALS determines absolute molar mass directly after chromatographic separation, distinguishing monomers from dimers and aggregates. AUC measures sedimentation coefficients and molecular weights under native conditions, providing association constants and stoichiometry. SAXS generates low-resolution three-dimensional envelopes and radii of gyration (Rg) in solution. Native MS directly observes intact oligomeric species and subunit composition. These methods collectively define the oligomeric state, association–dissociation equilibrium, and overall architecture of multi-subunit proteins.
Q: What is HDX-MS, and why is it valuable for HOS characterization?
A: Hydrogen–deuterium exchange mass spectrometry (HDX-MS) measures the rate at which backbone amide hydrogens exchange with deuterium in a D2O buffer. Residues in rigid, buried, or hydrogen-bonded regions exchange slowly, while those in flexible or solvent-exposed regions exchange rapidly. By digesting the protein into peptides and measuring deuterium uptake by LC-MS/MS, HDX-MS provides site-specific information on conformational dynamics, ligand-binding interfaces, and local stability. It is uniquely valuable because it bridges the gap between low-resolution global techniques (CD, DSC) and high-resolution static structures (X-ray, cryo-EM), offering dynamic, solution-phase data at peptide-level resolution.
Q: Can these methods detect subtle differences for biosimilar comparability?
A: Yes. The FDA and EMA biosimilar guidelines explicitly require demonstration of HOS similarity between a biosimilar candidate and its reference product. Our orthogonal platform—combining CD, FTIR, DSC, HDX-MS, and SEC-MALS—provides multiple independent assessments of structural similarity. Statistical algorithms (e.g., equivalence testing, PCA, and spectral overlay metrics) quantify the degree of similarity for each technique. HDX-MS is particularly powerful for biosimilarity because it can detect localized conformational differences that global methods might miss. Profacgen has supported numerous biosimilar programs with HOS packages accepted by major regulatory agencies.
Q: What sample requirements and turnaround times should we expect?
A: For spectroscopic methods (CD, FTIR, fluorescence), we typically require 0.5–2.0 mg of purified protein at ≥1 mg/mL in a buffer compatible with the technique (avoid high concentrations of histidine, imidazole, or detergents for CD). DSC requires 0.2–0.5 mg at 0.5–2.0 mg/mL. HDX-MS requires 50–100 µg per time point, with a typical experiment comprising 6–8 time points. SEC-MALS and AUC require 100–200 µg at 1–10 mg/mL. Standard turnaround is 10–15 business days for CD/FTIR/DSC, 15–20 business days for HDX-MS, and 10–14 business days for SEC-MALS/AUC. Rush services are available for time-critical regulatory submissions.
References:
Delfi M, Sartorius R, Ashrafizadeh M, et al. Self-assembled peptide and protein nanostructures for anti-cancer therapy: Targeted delivery, stimuli-responsive devices and immunotherapy. Nano Today. 2021;38:101119. doi:10.1016/j.nantod.2021.101119
Online Inquiry
Fill out this form and one of our experts will respond to you within one business day.