We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our
Privacy Policy
Profacgen's Process-Related Impurity Analysis service delivers comprehensive identification, quantification, and risk assessment of extrinsic contaminants introduced during upstream expression, downstream purification, and final formulation. From host cell proteins and residual DNA to chromatography resin leachables and solvent traces, we ensure your process-derived impurity profile meets the most stringent regulatory safety thresholds.
Process-related impurities are not merely analytical checkpoints—they directly impact patient safety, immunogenicity risk, and regulatory approval timelines. The ability to detect, clear, and document these residuals with platform-specific, phase-appropriate methods determines the robustness of your Chemistry, Manufacturing, and Controls (CMC) strategy and the speed of your path to clinic.
What Challenges Do We Solve?
Biopharmaceutical development teams face increasing pressure to resolve process-derived contamination with precision, sensitivity, and regulatory rigor. Key analytical and strategic challenges include:
Detecting and quantifying host cell proteins (HCPs) with sufficient proteome coverage and sensitivity to satisfy FDA and EMA expectations for clearance validation and immunogenicity risk assessment
Quantifying residual host cell DNA (HCD) down to picogram-per-dose levels to meet WHO safety limits and demonstrate robust nuclease and purification clearance
Validating Protein A ligand leaching from affinity chromatography resins and confirming clearance below safety thresholds for monoclonal antibody and Fc-fusion programs
Profiling culture media residuals, antifoam agents, buffer salts, organic solvents, and detergent traces that may persist through purification and formulation
Characterizing extractables and leachables from single-use bioreactor films, filtration membranes, and chromatography column packings under worst-case process conditions
Designing orthogonal clearance studies that demonstrate robust impurity removal across process changes, scale-up, site transfer, and post-approval modifications
Compiling regulatory-compliant data packages that satisfy ICH Q6B, ICH Q3B(R2), and agency-specific guidance for IND, BLA, and post-marketing submissions
Figure 1. Sources contributing to biopharmaceutical process-related impurities. (Geigert, 2019)
Addressing these challenges requires more than single-technique screening. It demands an integrated analytical architecture that connects physicochemical detection to biological risk assessment and regulatory strategy.
Profacgen's value drivers include:
Platform-specific method selection aligned with your expression system (CHO, E. coli, yeast, insect, or human) and purification train
Phase-appropriate validation from platform screening assays to fully qualified GMP release methods with pre-established acceptance criteria
Direct integration with upstream, downstream, and formulation development teams for real-time root-cause analysis and risk mitigation
Regulatory-ready documentation designed for seamless IND, BLA, and post-approval change management submissions
These priorities ensure your program advances with confidence, knowing that process safety is defined by data rather than assumption.
When to Consider Process-Related Impurity Analysis
Process-Related Impurity Analysis is most relevant when:
IND-enabling toxicology batches require comprehensive impurity profiling and safety qualification prior to first-in-human studies
Process characterization and validation studies must demonstrate clearance of HCP, DNA, and resin residuals with statistical confidence
Manufacturing scale-up or tech transfer requires rigorous comparability of impurity profiles across development sites and production scales
Post-approval process changes—such as resin replacement, filter change, or buffer modification—require re-validation of impurity clearance and regulatory filing
Biosimilar development programs must demonstrate equivalence of process-related impurity profiles against the reference product
Regulatory observations or complete response letters (CRLs) demand enhanced impurity investigation, method remediation, or additional clearance data
This service is particularly effective for teams that seek to de-risk CMC development by embedding impurity control into process design rather than treating it as a downstream analytical afterthought.
Our Core Platforms
Profacgen provides a structured, quality-oriented Process-Related Impurity Analysis service that aligns analytical methodology with your specific expression system, purification platform, and regulatory jurisdiction.
Our Residual Impurity Testing service delivers comprehensive profiling of process-derived chemicals, media components, antifoam agents, buffer salts, and solvent residuals. Using validated GC-MS, LC-MS/MS, and ion chromatography workflows, we detect and quantify trace-level residuals with sensitivity appropriate to your daily dose and administration route, ensuring compliance with ICH Q3C(R8) and Q3B(R2) safety thresholds.
The HCP Analysis service deploys orthogonal coverage-based methods including ELISA, mass spectrometry (LC-MS/MS), and Western blot to detect, identify, and quantify host cell proteins with broad proteome coverage. We support platform-specific polyclonal antibody generation, coverage analysis by 2D-DIBE or AEX/RP-HPLC, and risk-based HCP identification to ensure clearance validation meets regulatory expectations for immunogenicity risk assessment.
Our Residual DNA Quantification service utilizes quantitative PCR (qPCR), digital PCR (dPCR), and hybridization-based methods to detect host cell DNA with single-picogram sensitivity. Methods are validated for specificity, linearity, and precision across your cell line (CHO, E. coli, yeast, human) and purification process, supporting safety calculations that demonstrate residual DNA remains well below WHO-recommended limits of 10 ng per dose.
The Residual Protein A Analysis service targets leached Protein A ligand from affinity chromatography resins—a critical impurity for monoclonal antibody and Fc-fusion programs. We develop and validate sensitive ELISA and LC-MS/MS methods capable of detecting Protein A at sub-ng/mL levels, supporting clearance validation, in-process control, and release testing to ensure patient safety and regulatory compliance.
We conduct systematic risk assessments that map potential impurity sources to your specific process configuration, enabling proactive control strategy design rather than reactive troubleshooting.
Expression system-specific HCP risk profiling tailored to CHO, E. coli, yeast, insect, or human cell substrates
Downstream clearance factor analysis for each unit operation, from harvest and clarification through polishing and formulation
Leachable and extractable screening for single-use bioreactor films, tubing assemblies, and filtration devices under worst-case process conditions
Antibiotic, selection marker, and inducer residual assessment for compliant cell line systems
Viral clearance validation support through orthogonal impurity tracking and spiking studies
Regulatory & Documentation Support
IND-Enabling Packages: Platform method descriptions, preliminary acceptance criteria, and safety qualification summaries for Phase I/II submissions
BLA-Ready Validation: Full ICH Q2(R2) method validation with statistical analysis, system suitability criteria, and robustness demonstration for commercial release
Process Validation Support: Clearance factor documentation, spike-recovery studies, and orthogonal confirmation across scaled manufacturing batches
Post-Approval Change Management: Comparability protocols, risk assessments, and regulatory briefing documents for process changes, site transfers, and vendor switches
Regulatory-Centric Method Design: Every assay is architected with end-game filing requirements in mind, ensuring data packages withstand FDA, EMA, and NMPA scrutiny from IND through commercial license application.
Orthogonal Platform Breadth: No single technique captures the full spectrum of process-related impurities. Our multi-modal chromatography, mass spectrometry, electrophoresis, and molecular biology platforms eliminate analytical blind spots and build bulletproof comparability arguments.
Phase-Appropriate Agility: From early-stage platform screening to fully validated GMP release assays, we scale analytical rigor, system suitability criteria, and documentation to match your precise program milestone.
Integrated CMC Problem-Solving: Impurity data feeds directly into our upstream, downstream, and formulation teams, enabling rapid root-cause analysis, risk mitigation, and streamlined tech transfer to your manufacturing partner.
Application Scenarios
Scenario 1: HCP Clearance Validation for a Bispecific Antibody
Program Context:
A Phase I bispecific antibody program required demonstration of HCP clearance through a novel dual-affinity purification train, with regulators requesting coverage analysis beyond standard ELISA.
Objective:
Validate HCP clearance to below 100 ppm with orthogonal MS-based identification of any species present above 100 ppm for toxicological risk assessment.
Approach:
Profacgen deployed platform ELISA for total HCP quantification, followed by LC-MS/MS proteomics for species-specific identification, and 2D-DIBE coverage analysis of the anti-HCP polyclonal antibody. Spike-recovery studies confirmed clearance factors exceeding 10^4 across Protein A, AEX, and HIC steps.
Outcome:
The data package supported IND submission with zero regulatory queries on HCP, accelerated Phase I initiation by eight weeks, and established a platform method readily transferable to the client's CMO.
Scenario 2: Residual DNA Control in a Viral Vector Gene Therapy Program
Program Context:
An AAV gene therapy client faced heightened regulatory scrutiny on residual HEK293 DNA due to the high-dose regimen and intravitreal administration route, where standard WHO 10 ng/dose limits were considered insufficiently conservative.
Objective:
Develop a validated dPCR method with single-picogram sensitivity, and demonstrate process clearance to below 0.1 pg/dose—two orders of magnitude below conventional thresholds.
Approach:
Profacgen designed a dPCR assay targeting highly repetitive ALU elements for maximal sensitivity, executed across harvest, nuclease digestion, anion exchange, and ultrafiltration steps. Full ICH Q2(R2) validation including specificity against plasmid backbone DNA was completed.
Outcome:
The client received a complete validation report and clearance summary accepted by regulatory agencies, enabling Phase I/II trial initiation without holding toxicology batches for impurity retesting.
Q: What distinguishes process-related impurities from product-related impurities?
A: Process-related impurities are extrinsic entities introduced during manufacturing—host cell proteins, DNA, chromatography ligands, media components, solvents, and leachables. Product-related impurities are molecular variants of the drug substance itself, including aggregates, fragments, charge variants, and chemically modified species.
Q: Why is HCP analysis critical for biopharmaceutical safety?
A: Host cell proteins can trigger immunogenic responses, interfere with drug mechanism of action, or act as adjuvants that amplify immune reactions. Regulatory agencies require robust HCP detection, clearance validation, and risk assessment to ensure patient safety.
Q: What methods are used for residual DNA quantification?
A: We utilize quantitative PCR (qPCR), digital PCR (dPCR), and hybridization-based assays. dPCR offers absolute quantification without standard curve dependency, making it ideal for low-level residual DNA detection in gene therapy and high-dose biologics.
Q: How is Protein A clearance validated?
A: Protein A clearance is validated through sensitive ELISA or LC-MS/MS methods capable of sub-ng/mL detection. Spike-recovery studies across the purification train demonstrate clearance factors, and in-process controls ensure consistent removal batch-to-batch.
Q: What regulatory standards govern process-related impurity acceptance criteria?
A: Our methods align with ICH Q6B, ICH Q3B(R2), ICH Q5A(R2) for viral safety, WHO guidelines for residual DNA, and FDA guidance on immunogenicity assessment. Acceptance criteria are tailored to product class, clinical phase, route of administration, and dosing regimen.
Q: Can you support process validation and clearance factor studies?
A: Yes. We design and execute orthogonal clearance studies with spiking experiments, scale-down model qualification, and statistical analysis to demonstrate robust impurity removal. Deliverables include validation reports, SOPs, and regulatory briefing documents.
References:
Geigert J. Complex process-related impurity profiles. In: The Challenge of CMC Regulatory Compliance for Biopharmaceuticals. Springer International Publishing; 2019:231-260. doi:10.1007/978-3-030-13754-0_8
Online Inquiry
Fill out this form and one of our experts will respond to you within one business day.

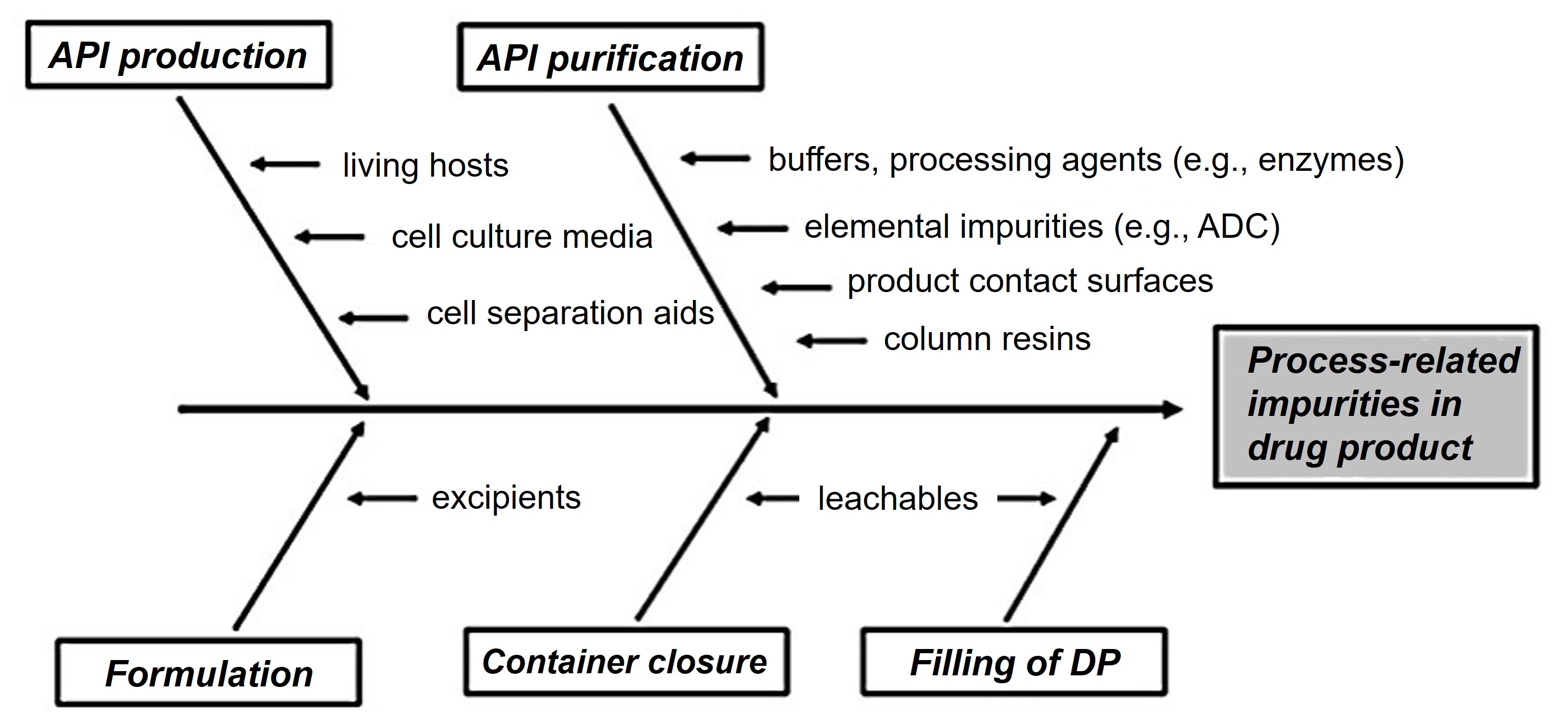 Figure 1. Sources contributing to biopharmaceutical process-related impurities. (Geigert, 2019)
Figure 1. Sources contributing to biopharmaceutical process-related impurities. (Geigert, 2019)