We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our
Privacy Policy
Profacgen's Comparability Studies service provides comprehensive, ICH Q5E-aligned analytical comparability assessment for biopharmaceutical products undergoing manufacturing changes, process scale-up, site transfer, or formulation modifications. Our programs demonstrate that post-change products maintain quality, safety, and efficacy profiles comparable to pre-change materials, supporting regulatory submissions and lifecycle management decisions.
Manufacturing changes are inevitable in the biopharmaceutical lifecycle. Process optimization, scale-up, technology transfer, and formulation revisions are essential for commercial viability and supply continuity. However, even seemingly minor modifications can alter product quality attributes with potential clinical implications. ICH Q5E establishes the framework for demonstrating that such changes do not adversely affect the product, but the analytical and regulatory complexity of comparability assessment is substantial. Profacgen provides the structured, science-driven approach required to navigate this complexity with confidence.
Introduction to Comparability Studies
Comparability is defined as the quality of being equivalent in all critical quality attributes that influence safety and efficacy. ICH Q5E (Quality of Biotechnological Products: Comparability of Biotechnological/Biological Products Subject to Changes in Their Manufacturing Process) provides the foundational guidance for demonstrating comparability, emphasizing that the goal is not identity but equivalence—no adverse impact on safety or efficacy.
The comparability exercise integrates analytical data, nonclinical information, and clinical experience to form a totality of evidence. When analytical comparability is comprehensively demonstrated, additional nonclinical or clinical studies may be reduced or eliminated. Conversely, analytical gaps or observed differences may necessitate targeted studies to address residual uncertainty. The quality of the analytical comparability package therefore directly influences the scope and cost of downstream development.
Key principles of comparability assessment include:
Quality attribute evaluation: Comprehensive comparison of physicochemical, biological, and immunochemical properties
Process understanding: Mechanistic knowledge of how changes may affect product quality
Risk-based approach: Focusing analytical effort on attributes most likely affected by the specific change
Statistical rigor: Objective comparison with predefined acceptance criteria and appropriate statistical methods
Totality of evidence: Integration of all available data to support the comparability conclusion
Figure 1. Assessed and assumed comparabilities for reference biologics and biosimilars. The figure shows the different evidentiary standards applied, without scientific basis, to reference products and biosimilars in geographic extensions. (Webster et al., 2021)
When Comparability Studies Are Required
Comparability studies are required across diverse scenarios in the biopharmaceutical lifecycle:
Manufacturing process changes: Cell culture media optimization, bioreactor parameter modification, purification resin replacement, or chromatography step resequencing
Scale-up: Transition from development-scale to pilot-scale or commercial-scale manufacturing with associated equipment and parameter changes
Site transfer: Technology transfer to new manufacturing facilities, CMOs, or geographic regions with different equipment, personnel, and environmental conditions
Formulation changes: Buffer modification, excipient substitution, concentration adjustment, or container-closure system replacement
Raw material changes: Supplier qualification, cell bank replacement, or critical reagent sourcing modifications
Post-approval changes: Process improvements, yield enhancements, or cost reduction initiatives requiring regulatory notification or approval
What We Offer
Comparability Strategy Design
Profacgen designs comparability strategies tailored to the specific change, product characteristics, and regulatory requirements:
Change impact assessment: Evaluation of the proposed modification against process understanding to identify potential quality attribute impacts
CQA risk ranking: Prioritization of attributes most likely affected by the change, focusing analytical resources on highest-risk areas
Study design and acceptance criteria: Definition of analytical panels, lot numbers, statistical methods, and predefined comparability margins
Regulatory pathway determination: Classification of change severity (minor, moderate, major) and corresponding submission requirements
Analytical Comparability Assessment
Comprehensive analytical comparison of pre-change and post-change products using orthogonal methods across structural, functional, and purity domains:
Structural comparability: Primary sequence confirmation, intact mass, peptide mapping, disulfide bond analysis, and higher-order structure assessment by CD and FTIR
Effector function comparability: ADCC, CDC, and complement activation assessment for antibody products
Stability Comparability
Comparison of degradation profiles and shelf-life performance under real-time and accelerated conditions:
Real-time stability comparison: side-by-side evaluation under intended storage conditions with identical pull points
Accelerated stress comparison: forced degradation under elevated temperature to compare degradation kinetics and pathways
Degradation product profiling: identification and quantification of species formed during stress to confirm equivalent degradation behavior
In-use stability comparability: evaluation of post-change product performance during clinical handling and administration
Risk Assessment & Data Interpretation
Profacgen provides comprehensive risk assessment and statistical interpretation that transforms analytical data into defensible comparability conclusions:
Statistical comparison methods: equivalence testing, quality range analysis, and descriptive comparison tailored to CQA criticality and data distribution
Multi-lot variability assessment: evaluation of pre-change and post-change batch-to-batch consistency to distinguish change effects from inherent variability
Residual uncertainty evaluation: systematic identification of analytical gaps or observed differences requiring additional study
Comparability conclusion documentation: formal assessment with scientific rationale supporting the determination of comparable or not comparable
Regulatory Support
Profacgen provides regulatory-compliant documentation and strategic support for comparability submissions:
Comparability protocols and reports: comprehensive documentation with predefined acceptance criteria, raw data, statistical analyses, and scientific conclusions
Regulatory submission packages: formatted for inclusion in IND amendments, BLA supplements, post-approval change management notifications, and MAA variations
Agency interaction support: preparation for scientific advice meetings, pre-submission conferences, and inspection readiness
Post-approval commitment management: execution of comparability commitments with defined timelines and reporting requirements
Specialized Service Modules
Profacgen's Comparability Studies service encompasses two integrated modules addressing distinct regulatory and analytical scenarios:
Structured comparability programs for process modifications, scale-up, technology transfer, and post-approval changes. Services include change impact assessment, CQA risk ranking, analytical comparability design, statistical comparison, and regulatory submission support aligned with ICH Q5E expectations for demonstrating equivalent quality, safety, and efficacy after manufacturing changes.
Focused analytical comparison of pre-change and post-change products with comprehensive structural, functional, purity, and stability assessment. Services include orthogonal method panels, multi-lot comparison, statistical equivalence evaluation, degradation pathway analysis, and integrated data interpretation supporting comparability conclusions for regulatory filing and quality system integration.
ICH Q5E Regulatory Alignment: Programs designed around ICH Q5E principles with risk-based CQA assessment, predefined comparability margins, and submission-ready documentation that satisfies FDA, EMA, and global regulatory expectations.
Change-Specific Strategy Design: Tailored comparability strategies based on mechanistic process understanding and change impact assessment, focusing analytical effort where risk is highest and avoiding unnecessary testing.
Orthogonal Analytical Depth: Multi-platform characterization spanning structural, functional, purity, and stability domains with independent methods that maximize confidence in comparability conclusions.
Statistical Rigor and Interpretation: Comprehensive data analytics including equivalence testing, variability assessment, and residual uncertainty evaluation that transform raw data into regulatorily defensible conclusions.
Integrated Lifecycle Support: Seamless coordination from change assessment through analytical comparability, regulatory submission, and post-approval commitment management within unified program oversight.
Post-Change Continuity Assurance: Bridging studies and stability comparability programs that demonstrate sustained equivalence over time, supporting uninterrupted commercial supply and patient access.
Representative Case Studies
Case 1: Process Scale-Up Comparability for Commercial Antibody
Background:
A commercially approved monoclonal antibody required manufacturing scale-up from 2,000 L to 10,000 L bioreactors with associated process parameter modifications. The change required demonstration of analytical comparability to support a post-approval change supplement and maintain uninterrupted commercial supply.
Our Solution:
Profacgen designed a risk-based comparability strategy focusing on attributes potentially affected by scale-up: glycosylation, aggregate content, charge variants, and potency. Ten pre-change lots and ten post-change lots were compared using orthogonal structural and functional methods. Statistical analysis employed equivalence testing for Tier 1 CQAs and quality range comparison for Tier 2 attributes. Forced degradation studies confirmed equivalent stability profiles.
Final Results:
All CQAs met predefined comparability criteria, with post-change lots falling within established pre-change variability ranges. Glycosylation profiles showed minor differences in high-mannose content that were demonstrated not to affect potency or pharmacokinetic performance. The comparability package successfully supported regulatory review of the manufacturing change within the planned submission timeline, allowing continued commercial supply without interruption.
Case 2: Site Transfer Comparability with Formulation Change
Background:
A therapeutic protein was being transferred to a new CMO with concurrent formulation buffer modification to improve stability. The combined changes required comprehensive comparability assessment to demonstrate that neither the manufacturing site change nor the formulation modification adversely affected product quality.
Our Solution:
Profacgen conducted a two-phase comparability study: Phase 1 isolated the site transfer effect by comparing material manufactured at the new site using the old formulation; Phase 2 evaluated the formulation change by comparing old and new formulations at the new site. Each phase employed full analytical panels with statistical comparison. Stability studies were conducted under real-time and accelerated conditions for both phases.
Final Results:
The site transfer demonstrated full comparability with no significant analytical differences observed between manufacturing locations. The formulation change resulted in improved stability while maintaining equivalent potency and purity profiles. This phased comparability strategy enabled clear differentiation of the impact associated with each individual change. The completed comparability package successfully supported global regulatory submissions for both the manufacturing transfer and formulation update.
Case 3: Post-Approval Purification Resin Replacement
Background:
A commercial biopharmaceutical product required replacement of a critical purification resin due to supplier discontinuation. The resin change had potential to affect impurity clearance, aggregate removal, and charge variant distribution, necessitating targeted comparability assessment.
Our Solution:
Profacgen focused the comparability strategy on purity and impurity attributes most likely affected by resin change: host cell protein clearance, aggregate content, charge variants, and process-related impurities. Eight pre-change and eight post-change lots were compared with enhanced sensitivity methods. In-process samples were analyzed to demonstrate equivalent clearance kinetics. Stability comparability confirmed no accelerated degradation with the new resin.
Final Results:
All purity and impurity attributes met comparability criteria with post-change lots showing equivalent or improved clearance. No new impurities were detected. The comparability package supported a minor change notification rather than a major supplement, reducing regulatory timeline from 12 months to 30 days. The resin replacement was implemented without supply disruption.
A: Comparability assesses whether a product remains equivalent after a manufacturing change within the same development program, using extensive prior knowledge and process understanding. Biosimilarity assesses whether a new product developed by a different manufacturer is highly similar to a reference product, with limited prior knowledge. Comparability relies on process understanding; biosimilarity requires comprehensive head-to-head characterization.
Q: How many lots are required for comparability assessment?
A: ICH Q5E recommends a sufficient number of lots to adequately assess variability, typically 8–12 lots per condition (pre-change and post-change). The exact number depends on product variability, change magnitude, and regulatory requirements. Profacgen conducts power analyses to determine statistically justified lot numbers for each comparability exercise.
Q: What are the key ICH Q5E principles for comparability?
A: ICH Q5E emphasizes that comparability does not require identity but rather equivalence in quality attributes affecting safety and efficacy. Key principles include: quality attribute comparison, process understanding, risk-based analytical focus, statistical rigor, and integration of all available data (analytical, nonclinical, clinical) into a totality of evidence.
Q: How are comparability acceptance criteria established?
A: Acceptance criteria are based on pre-change product variability, analytical method precision, and clinical relevance. For high-risk attributes, equivalence testing with predefined margins is employed. For moderate-risk attributes, quality ranges (mean ± 3 SD of pre-change lots) are used. Low-risk attributes are evaluated descriptively. Criteria are justified scientifically and documented in the comparability protocol.
Q: What happens if comparability is not demonstrated?
A: If comparability is not demonstrated, additional studies are required to address the observed differences. Options include: further analytical characterization to understand the difference, nonclinical studies to evaluate safety impact, clinical studies to confirm efficacy equivalence, or process modification to eliminate the difference. Profacgen provides strategic guidance on the most efficient pathway to resolve comparability gaps.
Q: Can comparability studies reduce clinical study requirements?
A: Yes. Comprehensive analytical comparability demonstrating equivalent quality attributes can support reduced or waived clinical studies, particularly for well-characterized products with established mechanisms of action. The extent of clinical reduction depends on the change magnitude, analytical confidence, and regulatory precedent. Profacgen designs comparability programs to maximize the analytical foundation supporting streamlined regulatory pathways.
Q: What regulatory submissions require comparability data?
A: Comparability data is required for post-approval changes (supplements, variations, notifications), manufacturing technology transfers, process scale-up, formulation changes, and site changes. The submission type depends on change severity: minor changes may require annual reporting; moderate changes require prior approval supplements or Type II variations; major changes require extensive documentation and regulatory review.
References:
ICH Q5E. Comparability of Biotechnological/Biological Products Subject to Changes in Their Manufacturing Process. International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use; 2004.
FDA Guidance for Industry. Comparability Protocols—Chemistry, Manufacturing, and Controls Information. U.S. Food and Drug Administration; 2003.
EMA. Guideline on Comparability after a Change in the Manufacturing Process. European Medicines Agency; 2012.
Webster CJ, George KL, Woollett GR. Comparability of biologics: global principles, evidentiary consistency and unrealized reliance. BioDrugs. 2021;35(4):379-387. doi:10.1007/s40259-021-00488-5
Online Inquiry
Fill out this form and one of our experts will respond to you within one business day.

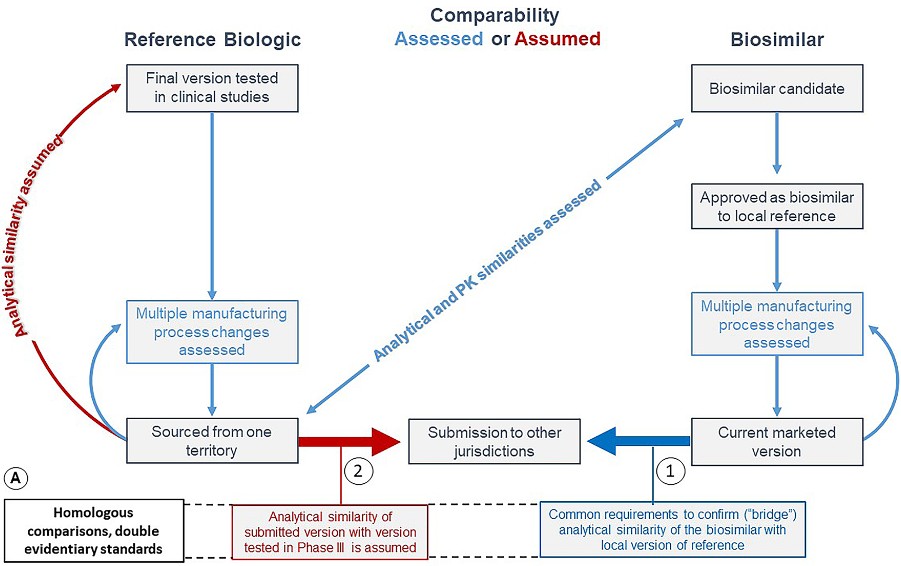 Figure 1. Assessed and assumed comparabilities for reference biologics and biosimilars. The figure shows the different evidentiary standards applied, without scientific basis, to reference products and biosimilars in geographic extensions. (Webster et al., 2021)
Figure 1. Assessed and assumed comparabilities for reference biologics and biosimilars. The figure shows the different evidentiary standards applied, without scientific basis, to reference products and biosimilars in geographic extensions. (Webster et al., 2021)