We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our
Privacy Policy
Phase-Appropriate Method Validation and Qualification
Phase-Appropriate Method Validation and Qualification
At Profacgen, our Phase-Appropriate Method Validation and Qualification service provides structured, risk-based analytical procedure validation aligned with program stage, regulatory expectations, and intended use. Our approach ensures that validation effort is proportionate to decision criticality, avoiding both under-validation that introduces compliance risk and over-validation that consumes unnecessary resources.
Validation represents the formal demonstration that an analytical method performs acceptably for its intended purpose. In biopharmaceutical development, where methods evolve from exploratory tools to regulatory submission pillars, the timing, scope, and rigor of validation must scale appropriately. Premature full validation of methods that may change wastes effort; delayed validation of methods supporting pivotal decisions introduces regulatory vulnerability. Profacgen's phase-appropriate framework resolves this tension through tiered validation strategies that match effort to risk.
Figure 1. Points to consider for performing validation readiness assessment.
Why Phase-Appropriate Validation Matters
Regulatory agencies and industry guidelines recognize that analytical methods at different development stages serve different purposes and therefore require different levels of formal demonstration. ICH Q2(R1) establishes the foundation for full validation, while FDA and EMA guidance acknowledge that early-phase methods may be qualified rather than fully validated, with validation completed prior to pivotal studies and commercial submission.
The consequences of misaligned validation strategy are significant. Under-validation at critical milestones can trigger regulatory holds, delay clinical trial initiation, or compromise submission readiness. Over-validation during exploratory phases diverts resources from higher-priority activities and may require repetition when methods evolve. A phase-appropriate approach optimizes resource allocation while maintaining compliance.
Key value drivers include:
Risk-based validation scope that focuses effort on methods supporting critical quality decisions
Regulatory alignment with FDA, EMA, and ICH expectations for each development phase
Efficient resource utilization by deferring full validation until method parameters are finalized
Seamless progression from qualification to validation without redundant experimentation
Documentation packages that support both immediate use and future regulatory submission
These priorities ensure that validation functions as a strategic enabler of efficient development rather than a procedural bottleneck.
Core Capabilities of Phase-Appropriate Validation
Profacgen provides a structured, quality-oriented Phase-Appropriate Method Validation and Qualification service that aligns validation scope with program stage, method criticality, and regulatory expectations.
Early-Phase Method Qualification
Formal demonstration of method suitability for IND-enabling and Phase I applications, with scope proportionate to early-stage decision needs. This includes:
Specificity and selectivity evaluation with representative samples and potential interferents
Preliminary precision and accuracy assessment to support sample analysis confidence
Linearity and range establishment adequate for anticipated sample concentrations
Documentation suitable for IND inclusion and regulatory inquiry response
Qualification provides sufficient evidence for early-phase use while preserving flexibility for method refinement as product knowledge evolves.
Full Method Validation for Pivotal and Commercial Programs
Comprehensive ICH Q2(R1) validation for methods supporting Phase II/III studies, BLA/NDA submission, and commercial release. This includes:
Specificity, linearity, range, accuracy, and precision (repeatability, intermediate precision, reproducibility)
Detection limit and quantitation limit determination for impurity methods
Robustness evaluation with deliberate parameter variation
Stability-indicating method verification with forced degradation samples
Validation protocols are designed with predefined acceptance criteria aligned with regulatory expectations and program-specific specifications.
Method Transfer Validation and Equivalence Studies
Validation support for method transfer between laboratories, CMOs, or geographic regions, demonstrating that receiving sites can execute methods with equivalent performance. This includes:
Comparative accuracy and precision evaluation between originating and receiving laboratories
Equivalence testing with statistical criteria (e.g., two one-sided t-tests for bias assessment)
Revalidation of modified methods when receiving site conditions necessitate adjustment
Transfer protocol and report documentation for regulatory filing and quality system integration
Transfer validation ensures method performance consistency across the manufacturing and testing network.
Post-Change Revalidation and Bridging Studies
Targeted validation or bridging studies following method modifications, process changes, or specification revisions. This includes:
Risk-based evaluation of change impact on method performance characteristics
Bridging study design focusing on affected validation parameters
Comparability assessment between pre-change and post-change method performance
Regulatory-compliant documentation supporting change control and submission amendments
Post-change validation is scoped to demonstrate that modified methods continue to meet intended purpose without redundant full revalidation.
Validation Parameters and Acceptance Criteria
Profacgen validates analytical methods with parameter selection and acceptance criteria tailored to method type and intended use:
Identification methods: specificity confirmed with representative samples, reference standards, and potential interferents; acceptance criteria based on peak resolution or spectral match thresholds
Impurity and purity methods: specificity, linearity, range, accuracy, precision, detection limit, and quantitation limit validated; acceptance criteria aligned with specification limits and ICH Q3B thresholds
Assay and potency methods: specificity, linearity, range, accuracy, precision, and robustness validated; acceptance criteria reflecting therapeutic potency range and clinical relevance
Biological methods: specificity, relative accuracy, precision, linearity, and range validated with appropriate statistical models; parallelism and system suitability criteria established
Risk-Based Scope Definition: Validation effort is proportionate to method criticality and program stage, avoiding both under-validation that invites regulatory challenge and over-validation that consumes resources without adding value.
Regulatory Phase Alignment: Validation strategies aligned with regulatory agencies’ expectations for each development phase, ensuring that qualification and validation packages meet reviewer expectations without unnecessary scope expansion.
Efficient Progression Design: Qualification studies are structured to support seamless progression to full validation, with early-phase data informing validation protocol design and reducing redundant experimentation.
Statistical Rigor: Validation data analyzed with appropriate statistical methods, including equivalence testing, confidence interval evaluation, and variance component analysis, ensuring that acceptance criteria are met with defensible quantitative evidence.
Change-Responsive Revalidation: Post-change validation and bridging studies are scoped based on structured risk assessment, focusing effort on affected parameters and avoiding unnecessary full revalidation.
Submission-Ready Documentation: Validation protocols, raw data, statistical analyses, and comprehensive reports prepared in formats suitable for direct regulatory submission, inspection presentation, and quality system archiving.
Representative Program Scenarios
Scenario 1: Phase-Appropriate Validation for Advancing Clinical Candidate
Program Context:
A therapeutic antibody program required analytical method support for Phase I IND submission, with subsequent progression to Phase III and BLA. The program needed early-phase qualification that would support initial clinical release without precluding efficient full validation for pivotal studies.
Objective:
To implement a tiered validation strategy with Phase I qualification of identity, purity, and potency methods, followed by full ICH Q2(R1) validation prior to Phase III initiation.
Approach:
Profacgen designed a phase-appropriate validation roadmap with defined qualification criteria for Phase I and full validation acceptance criteria for Phase III. Phase I qualification included specificity, preliminary precision, and linearity sufficient for clinical release and stability monitoring. Full validation expanded to include intermediate precision, robustness, and comprehensive accuracy assessment. Validation protocols were designed with predefined acceptance criteria and statistical analysis plans. Stability-indicating capability was verified with forced degradation samples during both phases.
Outcome:
Phase I qualification supported successful IND submission and clinical trial initiation without regulatory queries on analytical methods. Full validation was completed efficiently by leveraging qualification data, with all methods meeting acceptance criteria and supporting BLA submission. The tiered approach reduced total validation effort by approximately 30% compared to a non-strategic approach.
Scenario 2: Post-Approval Method Revalidation for Process Change
Program Context:
A commercially approved therapeutic protein underwent a manufacturing process change to improve yield and reduce impurities. The change affected the impurity profile, necessitating evaluation of whether existing purity methods remained suitable and required revalidation.
Objective:
To assess the impact of process change on method performance, design targeted revalidation studies, and demonstrate that methods continued to meet intended purpose with the modified process.
Approach:
Profacgen conducted a structured risk assessment evaluating the effect of process change on method specificity, accuracy, and precision. The assessment identified that the change introduced a new minor impurity species not resolved by the existing SEC method. A targeted revalidation focused on specificity enhancement through column and gradient optimization, followed by accuracy and precision re-evaluation with process change samples. Bridging data demonstrated equivalent performance for all previously validated parameters.
Outcome:
The targeted revalidation resolved the new impurity with baseline separation and demonstrated that all other method performance characteristics remained within acceptance criteria. The revalidation package supported the post-approval change supplement, and the modified method was implemented without disruption to commercial release testing.
Q: What is the difference between method qualification and full validation?
A: Method qualification provides formal evidence that a method is suitable for its intended use during early development, typically covering specificity, preliminary precision, and linearity. Full validation follows ICH Q2(R1) with comprehensive demonstration of all relevant performance characteristics including accuracy, precision (repeatability, intermediate, reproducibility), range, detection limit, and robustness. Qualification supports early-phase decisions; validation supports pivotal studies and commercial release.
Q: Which validation parameters are required for different method types?
A: ICH Q2(R1) defines validation parameter requirements by method category. Identification methods require specificity. Impurity quantitation methods require specificity, linearity, range, accuracy, precision, detection limit, and quantitation limit. Assay methods require specificity, linearity, range, accuracy, precision, and robustness. Biological assays require specificity, relative accuracy, precision, linearity, and range with appropriate statistical models.
Q: When should full validation be completed in a development program?
A: Full validation should be completed prior to initiation of pivotal Phase II/III studies to ensure that methods supporting primary efficacy and safety endpoints are fully demonstrated. Methods supporting commercial release and stability must be validated before BLA/NDA submission. Early-phase qualification may suffice for Phase I if methods are not yet finalized, with full validation completed during Phase II.
Q: What triggers method revalidation versus bridging studies?
A: Revalidation is required when method parameters are modified or when the method is applied to a fundamentally different matrix. Bridging studies are appropriate when process or formulation changes occur but method parameters remain unchanged, with focused evaluation of affected performance characteristics. Profacgen conducts risk-based assessments to determine the appropriate approach for each change scenario.
Q: How are acceptance criteria established for validation?
A: Acceptance criteria are established based on method intended use, specification requirements, regulatory expectations, and analytical capability. Criteria must be challenging yet achievable, with justification documented in the validation protocol. Profacgen establishes criteria through consultation with program teams, review of regulatory precedents, and assessment of method performance during development.
Q: Can qualification data be used to support full validation?
A: Yes, where qualification studies were designed with validation-compatible protocols and acceptance criteria, qualification data may be incorporated into full validation packages to reduce redundant experimentation. Profacgen structures early-phase qualification with this progression in mind, ensuring that data remain valid and defensible when included in subsequent validation documentation.
References:
ICH Q2(R1). Validation of Analytical Procedures: Text and Methodology. International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use; 2005.
ICH Q2(R2). Validation of Analytical Procedures. International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use; 2023.
FDA Guidance for Industry. Analytical Procedures and Methods Validation for Drugs and Biologics. U.S. Food and Drug Administration; 2015.
Online Inquiry
Fill out this form and one of our experts will respond to you within one business day.
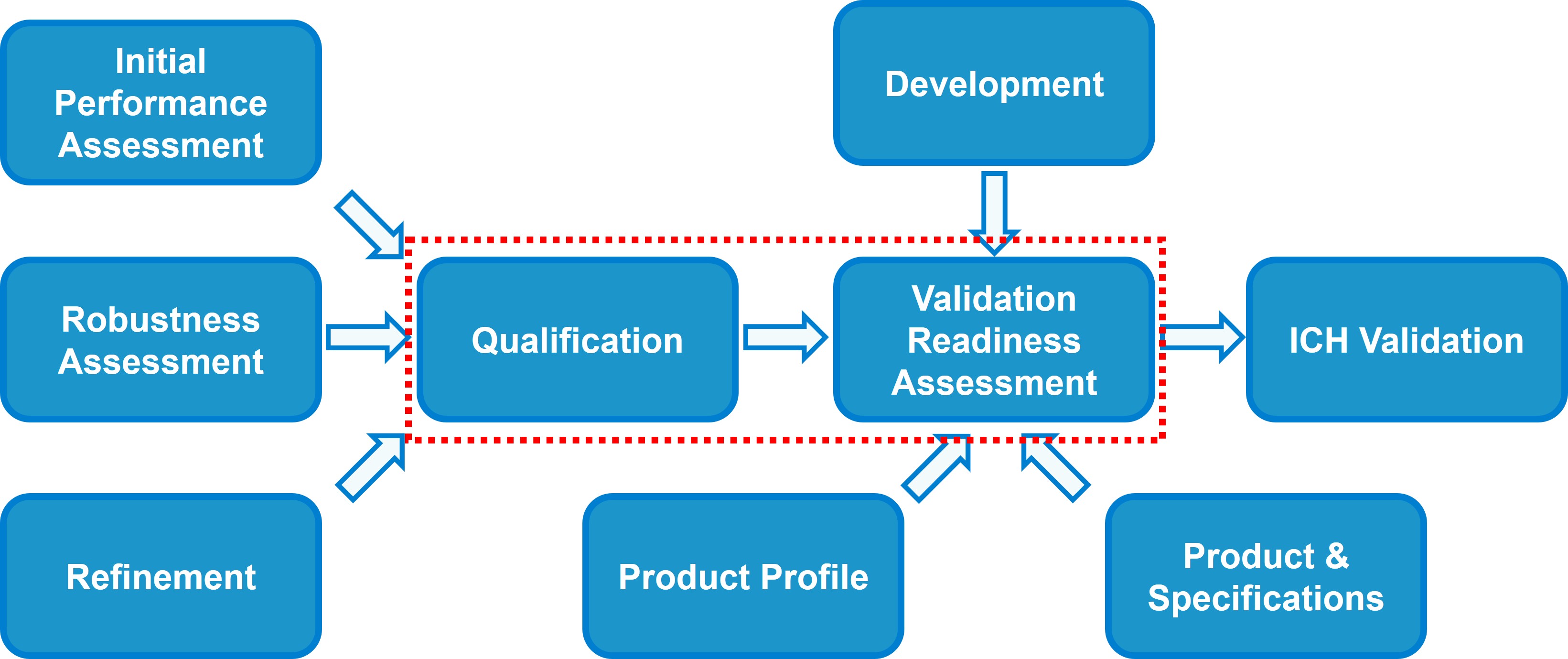 Figure 1. Points to consider for performing validation readiness assessment.
Figure 1. Points to consider for performing validation readiness assessment.