We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our
Privacy Policy
Protein A is a 42 kDa cell wall component of Staphylococcus aureus that binds with high affinity to the Fc region of immunoglobulin G (IgG). Owing to this specificity, immobilized Protein A and its engineered, alkali-stable recombinant variants are widely used as the industry standard for affinity capture of monoclonal antibodies (mAbs) and Fc-fusion proteins during downstream purification. However, leaching of Protein A ligand from chromatography resins into the drug substance poses potential safety risks. As a foreign protein, residual Protein A may trigger anti-drug antibody (ADA) responses, interfere with IgG-mediated effector functions, and exert immunomodulatory or mitogenic effects. In accordance with ICH Q6B and globally recognized regulatory expectations, manufacturers are required to demonstrate effective clearance of leached Protein A and to validate sensitive, reproducible analytical methods capable of detecting trace levels at the nanogram-per-milligram (ng/mg) or parts-per-million (ppm) range. Profacgen’s Residual Protein A Analysis platform provides integrated, phase-appropriate solutions that combine immunoassay-based quantification, mass spectrometry-driven identification, and process clearance evaluation to support comprehensive assessment of product purity and safety.
Figure 1. Staphylococcus aureus protein A bound to Fab and bound to an immunoglobulin Fc in its minimized form.
What Challenges Do We Solve?
Leached Protein A is one of the most stringently controlled process-related impurities in mAb manufacturing. Its structural similarity to the product and its potent immunomodulatory activity create unique analytical and regulatory challenges. Profacgen addresses the full spectrum of residual Protein A control:
Immunogenicity & Biological Activity Risks: Residual Protein A, as a foreign protein, may induce immune responses in varying degrees, bind circulating IgG via Fc interaction, and stimulate B-cell mitogenicity—necessitating rigorous control even at trace concentrations
Ultra-Low Detection Requirements: Regulatory expectations and industry practice typically demand Protein A levels below 10–20 ng/mg of product (10–20 ppm), requiring assays with exceptional sensitivity and minimal matrix interference from high-concentration antibody formulations
Product Interference & Specificity: The analyte (Protein A) and the product (IgG) share an Fc-binding interaction, creating a risk of co-precipitation, masking, or epitope competition that can compromise immunoassay accuracy
Ligand Diversity: Native S. aureus Protein A, recombinant Protein A, and engineered variants exhibit different epitope profiles, requiring assay customization or cross-reactivity validation for the exact resin employed
Process-Specific Leaching Profiles: Protein A leaching is influenced by resin age, cleaning-in-place (CIP) cycles, elution pH, and buffer composition; process changes require re-demonstration of clearance and may trigger method re-validation
Regulatory Documentation: ICH Q6B and FDA/EMA reviewers expect orthogonal evidence—combining immunoassay and mass spectrometry—that the purification process consistently clears leached ligand, supported by statistically robust specification justification
Our Core Platforms
Profacgen deploys a suite of orthogonal analytical platforms to detect, quantify, and characterize residual Protein A across the biopharmaceutical lifecycle. Each method is selected based on the resin chemistry, product concentration, matrix complexity, and regulatory phase, ensuring scientifically defensible and submission-ready data.
Platform
Capabilities & Deliverables
ELISA-Based Protein A Quantification
Development and ICH Q2(R1) validation of sandwich ELISA methods using anti-Protein A antibodies with demonstrated specificity for native, recombinant, or engineered ligand variants
High-throughput quantification with sensitivity down to low ng/mg (ppm) levels for routine release testing, stability monitoring, and in-process control
Matrix interference mitigation strategies (acid dissociation, chaotropic agents, or solid-phase extraction) to overcome IgG- Protein A complex formation in high-concentration formulations
LC-MS/MS Peptide Monitoring
Tryptic digestion followed by targeted multiple reaction monitoring (MRM) of Protein A-specific signature peptides, enabling antibody-independent quantification and orthogonal confirmation of ELISA results
Absolute quantification using stable isotope-labeled (SIL) peptide internal standards for accuracy and reproducibility in complex matrices
Identification of ligand degradation products and truncated leached species that may evade immunoassay detection
qPCR-Based Resin Gene Detection
Quantitative PCR targeting the spa gene or engineered recombinant sequences encoding the affinity ligand, applicable when the resin-derived protein is encoded by a known DNA construct
High sequence specificity and sensitivity for detecting residual genetic material associated with recombinant Protein A resins, complementing protein-level assays
Validation for specificity against host cell DNA and product-encoding sequences
SPR / BLI Biosensor Analysis
Surface plasmon resonance (SPR) and bio-layer interferometry (BLI) for real-time detection of Protein A-IgG interactions and free ligand quantification based on binding kinetics
Label-free detection with high sensitivity, useful for early-stage method feasibility, biosimilar comparability, and mechanistic studies of ligand-product interactions
Characterization of binding affinity differences between native and engineered Protein A variants
Process Clearance & Validation Studies
Controlled spiking studies across chromatographic and filtration steps to determine step-specific and cumulative Protein A clearance factors (purge factors)
Recovery validation and statistical process control to demonstrate consistent removal under routine and worst-case manufacturing conditions
Orthogonal Multi-Platform Portfolio: ELISA, LC-MS/MS, qPCR, and SPR/BLI are all available under one program, enabling cross-validated quantification and variant identification with consistent data interpretation.
Ligand Variant Differentiation: We distinguish between native S. aureus Protein A, recombinant forms, and engineered alkali-stable variants (e.g., MabSelect SuRe™), ensuring your assay detects the exact ligand chemistry used in your process.
Matrix Interference Resolution: Our optimized sample preparation protocols—including acid dissociation, solid-phase extraction, and chaotropic disruption—overcome Protein A-IgG complex formation and matrix suppression in high-concentration antibody formulations.
Regulatory-Ready Documentation: We deliver ICH Q2(R1) validation reports, method development narratives, clearance studies, specification justifications, and CTD-formatted control strategy documents for direct inclusion in IND, BLA, and MAA submissions.
Process Validation Integration: From resin qualification and spiking studies to statistical process control and purge-factor calculation, we provide the clearance evidence required to justify acceptance criteria and satisfy FDA/EMA expectations.
Phase-Appropriate Scalability: Whether you need a rapid ligand screen for early resin selection, a validated ELISA for Phase III release, or a comprehensive biosimilar similarity package, our modular service structure aligns with your development timeline.
Application Scenarios
Scenario 1: Commercial mAb Drug Substance Release & Stability
Challenge:
A commercial-stage manufacturer of a high-concentration (180 mg/mL) IgG1 monoclonal antibody observed sporadic out-of-specification (OOS) trends in residual Protein A during stability studies. The existing ELISA exhibited variable recovery, suspected to result from Protein A–IgG complex formation in the viscous, high-titer formulation matrix.
Our Approach:
We redeveloped the ELISA with an optimized acid-dissociation sample pretreatment step to disrupt Protein A–IgG complexes prior to analysis, followed by a validated sandwich format using a polyclonal anti-Protein A antibody with confirmed cross-reactivity against the client’s affinity ligand. Orthogonal LC-MS/MS targeting two signature tryptic peptides provided independent confirmation. The method was validated in accordance with ICH Q2(R1), with a demonstrated LOQ of 0.5 ng/mg and spike recovery of 85–115%.
Outcome:
The reformulated ELISA eliminated the OOS variability, and orthogonal LC-MS/MS confirmed that true Protein A levels remained consistently below 2 ng/mg across all stability time points. The updated method and validation package supported a successful post-approval change submission, enabling continued commercial supply without analytical concerns.
Scenario 2: Biosimilar Development & Analytical Similarity
Challenge:
A biosimilar developer needed to demonstrate analytical similarity between its candidate and the reference product with respect to residual Protein A. Industry expectations require comparable impurity profiles; however, differences in purification platforms, including the use of newer alkali-stable affinity resins, raised questions about whether distinct ligand chemistries could result in different leaching signatures.
Our Approach:
We deployed both ELISA (for total leached ligand quantification) and LC-MS/MS (for peptide-level variant identification) to characterize the residual Protein A profile. The LC-MS/MS panel distinguished between native Protein A domains and engineered ligand variants, confirming that the biosimilar process introduced a modified ligand population with distinct peptide signatures. Quantitative analysis demonstrated that total Protein A levels were statistically equivalent (mean difference <1 ng/mg), with no evidence of unique high-risk species.
Outcome:
The orthogonal similarity dataset provided robust support for comparability assessment. Equivalent total Protein A levels, combined with the absence of novel impurity species, established analytical similarity for this critical quality attribute and supported advancement through the development pathway.
Q: What is residual Protein A and why is it a critical process-related impurity?
A: Residual Protein A is the leached affinity ligand from Protein A chromatography resins used to capture monoclonal antibodies and Fc-fusion proteins. As a foreign protein derived from Staphylococcus aureus, it can induce anti-drug antibodies, bind circulating IgG via Fc interaction, and exert mitogenic or immunomodulatory effects. Regulatory authorities mandate strict control because even trace amounts pose immunogenicity and safety risks in patients.
Q: Which regulatory guidelines apply to residual Protein A testing?
A: ICH Q6B classifies leached ligands as process-related impurities requiring quantification and control. FDA and EMA guidance for monoclonal antibodies and biosimilars specify expectations for impurity profiling, including residual Protein A. ICH Q2(R1) governs the validation of analytical methods used for release and stability testing. Additionally, resin manufacturers provide leaching data, but sponsors must verify clearance under their specific process conditions.
Q: What are the typical acceptance criteria for residual Protein A in biopharmaceuticals?
A: Industry practice and regulatory precedent typically set residual Protein A limits at 10–20 ng per mg of product (10–20 ppm). Some high-risk products or pediatric indications may require more stringent criteria. Limits are justified based on clinical dosing, route of administration, immunogenicity risk assessment, and demonstrated process capability (statistical process control data).
Q: How does Profacgen overcome interference from the antibody product during Protein A analysis?
A: Because Protein A and IgG products bind via Fc interaction, complex formation can mask the analyte in immunoassays. We employ optimized sample pretreatment protocols—such as low-pH acid dissociation, chaotropic buffer disruption, or solid-phase extraction—to liberate free Protein A prior to quantification. These strategies are validated to ensure complete dissociation without analyte degradation, enabling accurate measurement even in high-concentration (≥150 mg/mL) formulations.
Q: Can Profacgen distinguish between different Protein A variants and engineered ligands?
A: Yes. We customize ELISA antibodies and LC-MS/MS peptide panels to detect native S. aureus Protein A, recombinant Protein A, and engineered variants such as MabSelect SuRe™ with modified IgG-binding domains. LC-MS/MS is particularly powerful for this application because it targets variant-specific amino acid sequences, enabling unambiguous identification of the exact leached ligand species.
Q: What is the difference between leached Protein A and other process-related impurities?
A: Leached Protein A is uniquely challenging because it is structurally and functionally related to the product (both bind IgG Fc), and it originates from a deliberate process contact material (the affinity resin) rather than the host cell. Unlike host cell proteins (HCPs) or residual DNA, Protein A is not an endogenous cellular component but an exogenous ligand introduced during purification. This distinction requires specialized analytical strategies to differentiate the ligand from the product and to validate clearance specifically attributable to the affinity step.
Q: What is the typical timeline for residual Protein A method development and validation?
A: ELISA method development and qualification typically require 4–6 weeks, including antibody selection, matrix interference mitigation, and preliminary validation. LC-MS/MS method setup adds 3–5 weeks for signature peptide selection and SIL internal standard synthesis. Full ICH Q2(R1) validation requires an additional 4–6 weeks. Integrated programs with parallel clearance study design can deliver a complete analytical and process validation package in 10–14 weeks, with expedited options for critical-path submissions.
Online Inquiry
Fill out this form and one of our experts will respond to you within one business day.
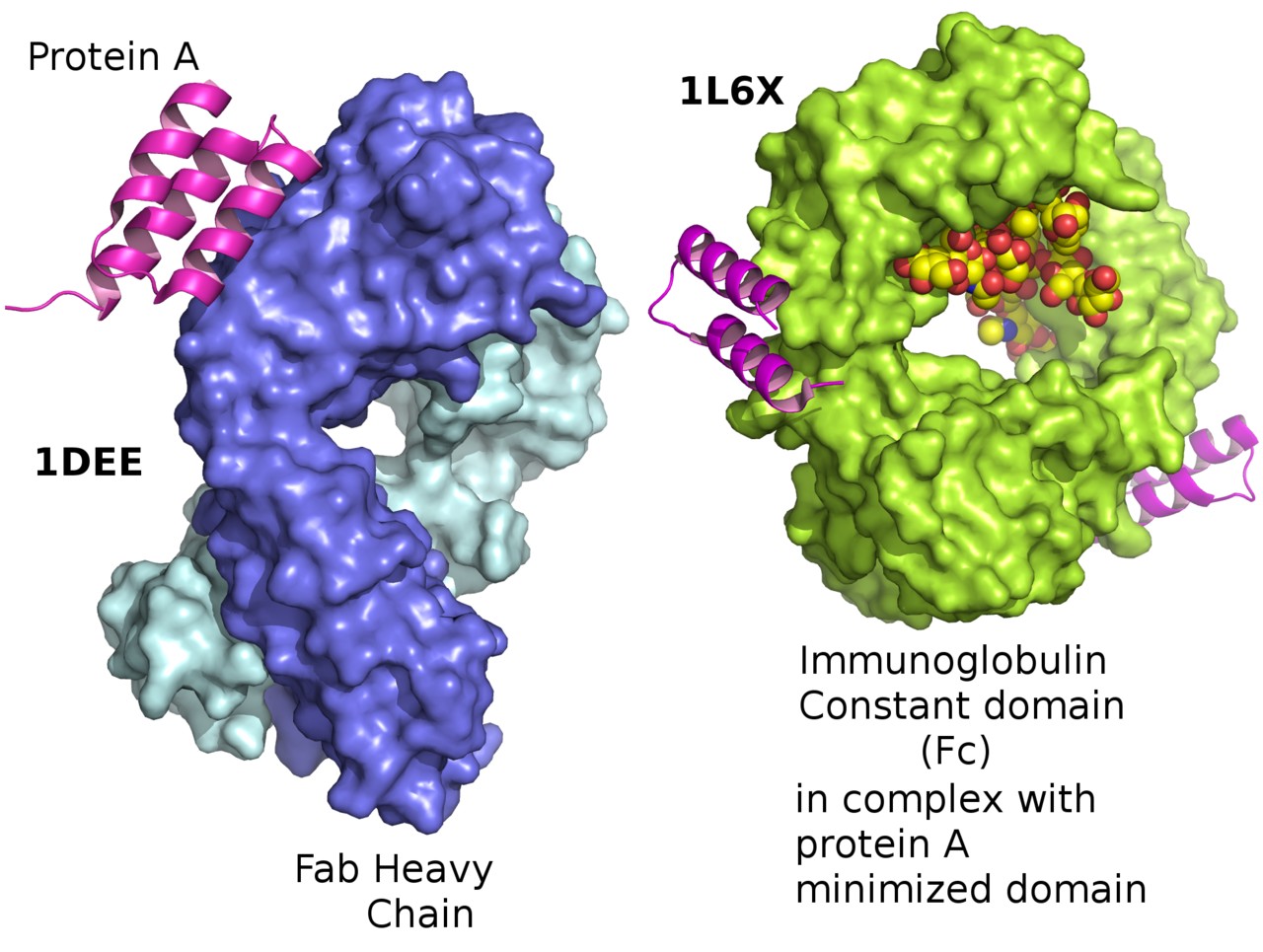 Figure 1. Staphylococcus aureus protein A bound to Fab and bound to an immunoglobulin Fc in its minimized form.
Figure 1. Staphylococcus aureus protein A bound to Fab and bound to an immunoglobulin Fc in its minimized form.