We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our
Privacy Policy
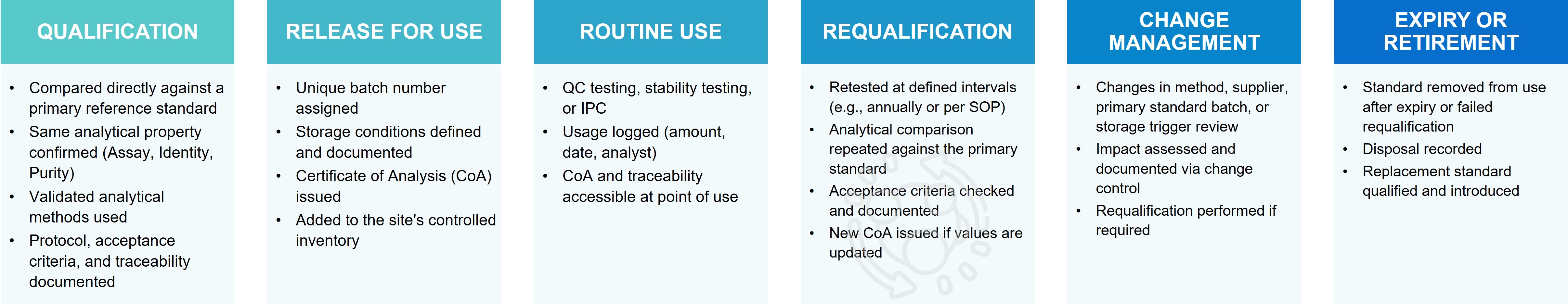
Profacgen's Reference Standard Requalification Programs provide periodic analytical verification that reference standards maintain assigned quality attributes throughout their designated shelf life. Our programs combine scheduled testing, stability trending, and degradation assessment to ensure that standards remain fit-for-purpose and that analytical continuity is preserved across lot transitions.
Reference standards degrade over time, even under optimal storage conditions. Proteins oxidize, aggregates form, and potency drifts gradually—changes that may not be immediately apparent but progressively compromise analytical reliability. Without systematic requalification, these changes accumulate undetected until a critical failure occurs: an out-of-trend result, a failed system suitability, or a batch release delay traced to standard degradation. Requalification is the proactive safeguard against this silent erosion of analytical quality.
Background: Why Reference Standard Requalification?
Regulatory expectations mandate that reference standards be re-evaluated periodically to confirm continued suitability. USP General Chapter <11> requires that standards be used within established periods, with evidence supporting assigned expiration dates. ICH Q6B emphasizes that specifications must be based on reference materials with demonstrated stability. These expectations are enforced through regulatory inspections, where requalification program deficiencies frequently generate observations affecting data integrity and batch release reliability.
The requalification challenge is timing: too frequent testing wastes resources and accelerates standard depletion; too infrequent testing risks undetected degradation compromising analytical results. Profacgen's programs employ risk-based interval determination, statistical trending, and predictive modeling to optimize testing frequency while maintaining confidence in standard suitability.
Key advantages of structured requalification include:
Early detection of degradation before analytical bias affects release decisions
Data-driven replacement timing preventing both premature disposal and overdue use
Regulatory compliance with documented requalification schedules and acceptance criteria
Bridging study support ensuring analytical continuity across standard lot transitions
Reduced OOS investigations by eliminating standard degradation as a root cause
Our Reference Standard Requalification Service Offerings
Profacgen provides tiered requalification programs tailored to standard type, stability knowledge, and program criticality.
Service Component
Description
Scheduled Requalification Testing
Risk-based interval determination using accelerated stability data and degradation kinetics
Comprehensive testing panels including identity, purity, and potency at defined time points
Statistical trending with control charts and regression analysis for degradation rate estimation
Formal requalification reports with pass/fail disposition and continued use authorization
Accelerated Stability Studies
Elevated temperature and stress condition evaluation to predict real-time degradation behavior
Arrhenius modeling for activation energy determination and shelf-life extrapolation
Degradation product profiling identifying primary and secondary degradation pathways
Support for shelf-life extension justifications and early replacement decisions
Replacement Strategy and Bridging Studies
New lot sourcing, characterization, and qualification with calibration against outgoing standard
Bridging study design demonstrating analytical equivalence between old and new lots
Statistical evaluation of potency ratio, purity profile, and performance consistency
Documentation supporting seamless transition without analytical discontinuity
Post-Excursion Requalification
Rapid requalification testing following storage temperature excursions or environmental deviations
Focused testing on parameters most sensitive to the specific stress condition
Impact assessment with documented rationale for release, restricted use, or destruction
Emergency response protocols with expedited turnaround for critical standards
Requalification Program Design and Documentation
Program protocol development with predefined testing intervals, acceptance criteria, and escalation pathways
Integration with inventory management systems for automated scheduling and notification
Annual program review with performance metrics, interval optimization, and regulatory compliance assessment
Comprehensive documentation packages for quality system integration and inspection readiness
Risk-Based Interval Optimization: Testing frequency is determined by degradation kinetics, accelerated stability data, and analytical criticality rather than arbitrary calendar schedules, optimizing resource utilization while maintaining confidence in standard suitability.
Predictive Degradation Modeling: Statistical trend analysis and Arrhenius modeling predict remaining standard lifetime, enabling proactive replacement planning and preventing emergency standard sourcing that disrupts analytical programs.
Seamless Lot Transition: Bridging studies with rigorous statistical equivalence evaluation ensure that new standard lots maintain analytical continuity, preserving trend consistency in stability data and release testing history across replacements.
Expedited Excursion Response: Post-deviation requalification protocols with focused testing parameters and rapid turnaround minimize standard downtime, supporting uninterrupted release testing and supply continuity after environmental stress events.
Regulatory Compliance Assurance: Programs aligned with USP <11>, ICH Q6B, and FDA expectations, with documented schedules, acceptance criteria, and formal reports that withstand inspection scrutiny and support submission requirements.
Integrated Program Automation: Requalification schedules interface with inventory and storage systems to trigger automated testing notifications, track standard consumption against remaining qualified lifetime, and generate compliance reports without manual intervention.
Representative Case Studies
Case 1: Risk-Based Interval Optimization for Commercial Standard
Background:
A commercial therapeutic protein reference standard was being requalified quarterly based on a conservative protocol established during early development. The frequent testing consumed 15% of total standard inventory annually and generated substantial analytical workload without evidence of degradation between tests.
Our Solution:
Profacgen analyzed 36 months of requalification data showing minimal potency drift (0.3% per year) and stable purity profiles. Accelerated stability studies at 25°C and 40°C confirmed low degradation activation energy. A risk-based interval extension to annual testing was proposed with statistical justification, supported by tightened alert limits for intermediate monitoring.
Final Results:
Annual requalification was approved by quality assurance and accepted during the subsequent inspection by the regulatory agencies. Standard consumption for requalification decreased by 70%, extending the effective standard lifetime. No degradation was detected over 24 months of extended-interval monitoring, validating the risk-based approach.
Case 2: Bridging Study Supporting Global Standard Harmonization
Background:
A biopharmaceutical company with regional manufacturing sites maintained separate working reference standards at each location, leading to potency assignment inconsistencies and analytical variability in inter-site comparability studies. Harmonization required replacing all regional standards with a single global lot.
Our Solution:
Profacgen qualified a new global standard with comprehensive characterization and potency assignment. A bridging study compared the new lot against all outgoing regional standards using the validated potency assay with multiple determinations per site. Statistical analysis evaluated potency ratios, precision, and equivalence across the transition.
Final Results:
The bridging study demonstrated equivalent potency ratios within ±2.0% across all sites, supporting seamless standard replacement. Inter-site analytical variability decreased by 40% post-harmonization. The global standard remains in use with consistent performance across the manufacturing network.
Q: How is requalification testing interval determined?
A: Intervals are determined by risk-based assessment incorporating accelerated stability data, real-time degradation trends, analytical criticality, and regulatory expectations. Standard programs may specify annual testing, while risk-optimized programs extend intervals when degradation kinetics support longer periods with maintained confidence.
Q: What testing is included in a typical requalification panel?
A: Typical panels include identity confirmation, purity assessment (aggregates, fragments, charge variants), potency determination, and subvisible particle analysis. The specific scope is tailored to the standard type, known degradation pathways, and regulatory requirements, with focused testing for post-excursion requalification.
Q: How are bridging studies designed for standard replacement?
A: Bridging studies compare the new standard against the outgoing lot using validated analytical methods with multiple independent determinations. Statistical evaluation includes potency ratio assessment, precision comparison, and equivalence testing to demonstrate that the transition maintains analytical continuity without systematic bias.
Q: Can shelf life be extended based on requalification data?
A: Yes, shelf life extensions may be supported when accumulated requalification data demonstrate stable performance beyond the original expiration date. Extensions require statistical trend analysis, degradation rate modeling, and documented justification reviewed and approved through formal change control procedures.
Q: What triggers immediate requalification outside scheduled intervals?
A: Immediate requalification is triggered by storage temperature excursions, visible physical changes, unexpected analytical trends suggesting degradation, or process changes affecting the analytical relationship between standard and sample. Post-excursion testing is typically expedited with focused parameters to minimize standard downtime.
Q: How does requalification integrate with inventory management?
A: Requalification schedules are integrated with inventory systems to trigger automated testing notifications, track consumption against remaining qualified lifetime, and coordinate replacement sourcing before expiry. This integration prevents use of expired standards and ensures continuous availability of qualified materials.
Q: What documentation is generated for regulatory compliance?
A: Documentation includes requalification protocols with acceptance criteria, analytical data reports, statistical trend analyses, formal disposition records, and program review summaries. For bridging studies, equivalence evaluation reports and potency ratio justifications are provided. All documentation supports inspection readiness and submission requirements.
References:
United States Pharmacopeia. USP General Chapter <11>: USP Reference Standards. United States Pharmacopeial Convention; current edition.
ICH Q6B. Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products. International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use; 1999.
United States Pharmacopeia. USP General Chapter <1041>: Biologics. United States Pharmacopeial Convention; current edition.
Online Inquiry
Fill out this form and one of our experts will respond to you within one business day.