We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our
Privacy Policy
Profacgen's GMP Lot Release Testing service provides comprehensive, regulatory-compliant analytical testing and quality evaluation for biopharmaceutical products manufactured under Good Manufacturing Practice conditions. Our testing programs are designed to verify that each manufactured lot meets predefined quality specifications prior to distribution, ensuring patient safety, regulatory compliance, and supply chain integrity.
Lot release testing represents the final quality gate before a biopharmaceutical product enters clinical or commercial distribution. The complexity of protein therapeutics—combined with the stringent expectations of global regulatory agencies—demands a testing partner with deep analytical expertise, GMP-aligned quality systems, and the ability to execute under tight timelines without compromising data integrity or scientific rigor.
Why GMP Lot Release Testing Matters
GMP lot release testing serves as the definitive verification that a manufactured batch conforms to the approved specifications established during development and validated during process qualification. For biopharmaceutical products, this encompasses a broad spectrum of quality attributes that reflect molecular identity, purity, potency, safety, and consistency.
The consequences of inadequate or delayed lot release testing extend beyond regulatory non-compliance. Release delays can disrupt clinical trial supply chains, compromise patient access to critical therapies, and incur substantial financial penalties. Conversely, robust testing programs with efficient turnaround times enable predictable supply, support just-in-time manufacturing strategies, and build regulatory confidence through consistent data quality.
Key value drivers include:
Regulatory-compliant testing under documented GMP quality systems
Rapid turnaround to support clinical and commercial supply schedules
Comprehensive analytical coverage spanning identity, purity, potency, and safety
Integrated data management and documentation for direct regulatory submission
Scalable capacity to accommodate program growth from clinical phases to commercial launch
These priorities ensure that lot release testing functions not merely as a compliance checkpoint, but as a strategic enabler of efficient, reliable biopharmaceutical supply.
When to Consider GMP Lot Release Testing
GMP Lot Release Testing is most relevant when:
Your program has advanced to GMP manufacturing and requires release testing for clinical trial material or commercial product
You need a testing partner with validated analytical methods, qualified instruments, and documented GMP quality systems
Regulatory submissions (IND, BLA, NDA, MAA) require comprehensive lot release data packages
Internal analytical capacity is insufficient to meet release timelines or regulatory expectations
This service is particularly effective for programs transitioning from development to regulated manufacturing, where the shift from research-grade to GMP-compliant testing represents a significant operational and quality systems challenge.
Core Capabilities of GMP Lot Release Testing
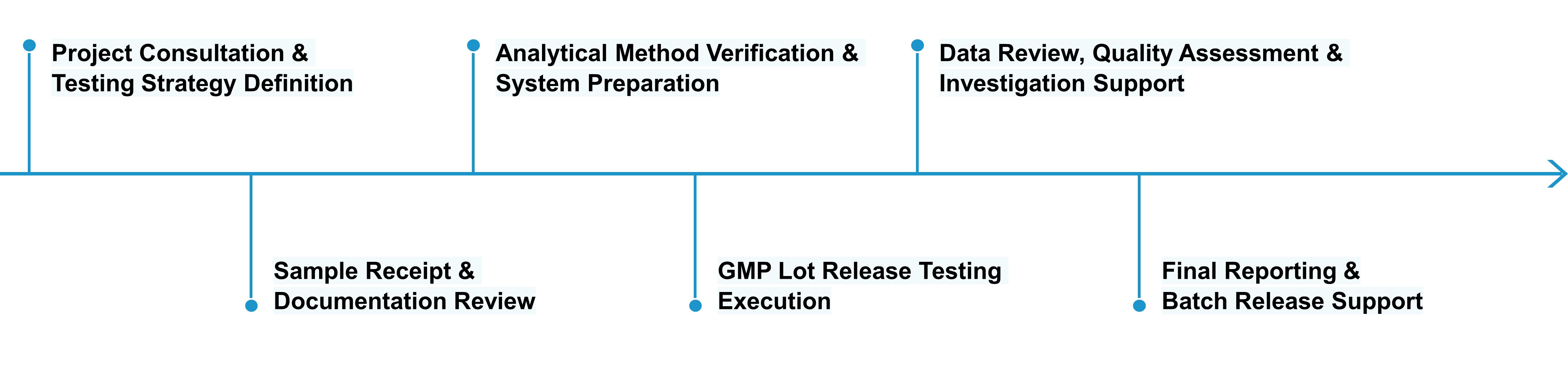
Profacgen provides a structured, quality-oriented GMP Lot Release Testing service that aligns analytical execution with regulatory requirements, manufacturing schedules, and program-specific quality specifications.
Identity and Purity Testing
Comprehensive verification of molecular identity and purity using validated analytical methods that confirm the product matches the approved reference standard and meets purity specifications. Capabilities include:
Peptide mapping with LC-MS/MS for primary sequence confirmation
Intact mass analysis by high-resolution mass spectrometry
Size exclusion chromatography (SEC-HPLC) for aggregate and fragment quantification
Capillary electrophoresis (CE-SDS) for reduced and non-reduced purity assessment
Ion-exchange chromatography (IEX) and imaged capillary isoelectric focusing (icIEF) for charge variant profiling
Each method is validated for specificity, precision, accuracy, and robustness under GMP conditions, with documented system suitability criteria and acceptance limits.
Potency and Biological Activity Assessment
Functional evaluation of therapeutic activity using validated bioassays that reflect the mechanism of action and clinical relevance of the product. Capabilities include:
Cell-based assays for receptor binding, signaling, or functional response
Enzymatic activity assays for catalytic therapeutics
Reporter gene assays for pathway-specific activity measurement
Complement-dependent cytotoxicity (CDC) or antibody-dependent cellular cytotoxicity (ADCC) assays for effector function evaluation
Reference standard calibration and relative potency determination with statistical validation
Bioassays are developed, qualified, and validated with appropriate controls, parallelism testing, and system suitability to ensure reproducible, decision-relevant potency data.
Safety and Microbiological Testing
Comprehensive safety testing to verify that the product is free from adventitious agents, endotoxins, and microbial contamination. Capabilities include:
Endotoxin testing by Limulus Amebocyte Lysate (LAL) assay or recombinant Factor C (rFC) methods
Sterility testing by membrane filtration or direct inoculation per USP <71>
Bioburden assessment by membrane filtration or pour plate methods
Mycoplasma detection by PCR-based or culture-based methods
Adventitious agent screening by in vitro and in vivo assays where required
Safety testing is conducted under documented aseptic conditions with appropriate environmental monitoring and controls to ensure result integrity and regulatory compliance.
Container-Closure and General Quality Testing
Physical and chemical testing to verify container-closure integrity, appearance, and general product quality. Capabilities include:
Visual inspection for particulates, color, clarity, and container defects
pH, osmolality, and conductivity measurement
Extractable volume and deliverable volume verification
Container-closure integrity testing by dye ingress, vacuum decay, or headspace analysis
Seal integrity and residual seal force evaluation
General quality testing ensures that the product meets all physical and chemical specifications, supporting comprehensive lot release decisions and patient safety.
GMP Quality Systems and Documentation
Profacgen's GMP Lot Release Testing operations are supported by comprehensive quality systems designed to meet global regulatory expectations:
Method validation and transfer: analytical methods are validated in accordance with ICH Q2(R1) guidelines, with documented validation protocols, reports, and ongoing method performance monitoring
Instrument qualification and calibration: all analytical instruments are qualified (IQ/OQ/PQ), calibrated against traceable standards, and maintained under documented preventive maintenance programs
Data integrity and ALCOA+ principles: electronic and paper-based data are managed in accordance with ALCOA+ principles (attributable, legible, contemporaneous, original, accurate, plus complete, consistent, enduring, available)
Change control and deviation management: all changes to methods, specifications, or processes are evaluated through formal change control, with deviations investigated, documented, and resolved through CAPA systems
Training and personnel qualification: testing personnel are trained and qualified on specific methods and instruments, with ongoing competency assessment and documentation
GMP-Aligned Quality Systems: Testing operations are conducted under documented GMP quality frameworks with full traceability, change control, and audit readiness, ensuring that generated data meet global regulatory expectations without additional qualification or remediation.
Comprehensive Analytical Coverage: Integrated testing across identity, purity, potency, safety, and general quality attributes within a single service provider, reducing program complexity, transfer risks, and timeline fragmentation associated with multiple vendors.
Method Validation Expertise: Deep experience in developing, qualifying, and validating analytical methods specifically for biopharmaceutical lot release, with documented validation packages that support regulatory submissions and post-approval compliance.
Scalable and Timely Execution: Flexible capacity and structured workflows that accommodate fluctuating lot volumes from clinical phases through commercial launch, with turnaround times optimized to support manufacturing and supply chain schedules.
Regulatory Documentation for Direct Submission: Comprehensive test reports, certificates of analysis, and supporting documentation prepared in formats suitable for direct inclusion in regulatory submissions, quality systems, and regulatory inspections.
Integrated Program Support: Seamless coordination with manufacturing, stability, characterization, and regulatory strategy teams to ensure that lot release data inform broader CMC decisions and maintain consistency across the product lifecycle.
Representative Program Scenarios
Scenario 1: Phase III Clinical Trial Material Release Testing
Program Context:
A biopharmaceutical company advancing a monoclonal antibody therapeutic into Phase III clinical trials required GMP-compliant lot release testing for multiple batches of clinical trial material manufactured at a CMO. The program needed rapid turnaround to maintain clinical supply continuity while ensuring full regulatory compliance for the upcoming BLA submission.
Objective:
To establish a GMP lot release testing program with validated methods, documented quality systems, and efficient execution that could support Phase III clinical supply and generate data packages suitable for BLA inclusion.
Approach:
Profacgen implemented a comprehensive lot release testing program encompassing identity (peptide mapping, intact mass), purity (SEC-HPLC, CE-SDS, icIEF), potency (cell-based binding assay, ADCC assay), safety (LAL endotoxin, sterility, bioburden), and general quality (appearance, pH, osmolality, container-closure integrity). All methods were validated under GMP conditions with documented validation protocols and reports. A dedicated project team was assigned to ensure rapid sample receipt-to-report turnaround, with electronic data management supporting real-time status tracking. Certificates of analysis were generated in a format aligned with the client's quality system and regulatory submission requirements.
Outcome:
The lot release testing program supported continuous clinical supply over 18 months with 100% on-time release and zero regulatory observations. The comprehensive validation packages and test reports were integrated directly into the BLA submission, contributing to a successful regulatory review and approval. The established testing framework was transitioned seamlessly to commercial lot release post-approval.
Scenario 2: Post-Approval Method Transfer and Release Testing Scale-Up
Program Context:
A commercially approved therapeutic protein faced manufacturing scale-up from single-site to multi-site production, requiring analytical method transfer, additional method validation, and expanded lot release testing capacity to support increased batch volumes and new manufacturing locations.
Objective:
To transfer validated lot release methods to Profacgen's GMP laboratory, execute method equivalence studies, and establish a scalable testing program capable of supporting doubled batch volumes without compromising turnaround times or data quality.
Approach:
Profacgen conducted a structured method transfer program including pre-transfer risk assessment, side-by-side equivalence testing with the originating laboratory, and post-transfer method performance qualification. Method validation was updated to reflect the new laboratory environment and expanded specification ranges associated with scale-up. A dedicated testing schedule was established with pre-aligned capacity allocation, enabling concurrent release testing for multiple lots. Electronic data management and automated reporting streamlined the certificate of analysis generation process.
Outcome:
The method transfer was completed with demonstrated equivalence across all critical quality attributes, and the expanded testing program supported a 100% increase in batch volume with maintained turnaround times. The program didn’t receive any inspection observations from regulatory agencies related to lot release testing during the subsequent PAI, and the client achieved an uninterrupted commercial supply throughout the scale-up transition.
Q: What distinguishes GMP lot release testing from research-grade analytical testing?
A: GMP lot release testing is conducted under documented Good Manufacturing Practice quality systems with validated methods, qualified instruments, trained personnel, and comprehensive data integrity controls. Test results are reported on official certificates of analysis suitable for regulatory submission and batch release decisions. Research-grade testing, while scientifically rigorous, lacks the formal quality system infrastructure, method validation documentation, and regulatory defensibility required for commercial batch release.
Q: Which analytical methods are typically included in a biopharmaceutical lot release panel?
A: A typical lot release panel for monoclonal antibodies or therapeutic proteins includes identity testing (peptide mapping, intact mass), purity assessment (SEC-HPLC, CE-SDS, icIEF), potency determination (cell-based or binding assays), safety testing (endotoxin, sterility, bioburden), and general quality tests (appearance, pH, osmolality). The specific panel is customized based on the product, regulatory requirements, and approved specifications.
Q: How long does GMP lot release testing typically take?
A: Turnaround times depend on the complexity of the analytical panel, method readiness, and sample volume. A standard lot release panel for a monoclonal antibody can typically be completed within 5–10 business days, with expedited timelines available for critical clinical supply needs. Sterility testing requires 14 days per pharmacopeial requirements. Profacgen works with clients to establish timelines that align with manufacturing and supply chain schedules.
Q: Can existing analytical methods be transferred to Profacgen for lot release testing?
A: Yes. Profacgen conducts structured method transfer programs in accordance with ICH Q2(R1) and industry best practices, including pre-transfer risk assessment, equivalence testing, and post-transfer performance qualification. We have extensive experience transferring methods from client laboratories, CMOs, and other testing providers, with documented transfer protocols and reports suitable for regulatory filing and quality system integration.
Q: What regulatory guidelines govern GMP lot release testing?
A: GMP lot release testing is governed by ICH Q7 (Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients), ICH Q10 (Pharmaceutical Quality System), regional GMP regulations (21 CFR Parts 210–211 in the United States, EudraLex Volume 4 in the EU), and pharmacopeial requirements (USP, EP, JP). Analytical method validation follows ICH Q2(R1), while specific tests such as sterility and endotoxin testing comply with USP <71>, USP <85>, and harmonized pharmacopeial chapters.
Q: How does Profacgen ensure data integrity in lot release testing?
A: Data integrity is ensured through adherence to ALCOA+ principles across all testing operations. This includes attributable electronic records with user authentication, audit trails for all data modifications, contemporaneous data capture, original record retention, and accurate transcription verification. Instruments are connected to validated data acquisition systems where possible, and manual entries are subject to independent verification. Regular data integrity audits and training programs reinforce compliance across all personnel.
Online Inquiry
Fill out this form and one of our experts will respond to you within one business day.