We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our
Privacy Policy
Cytochrome P450 (CYP) enzymes constitute the predominant system responsible for the oxidative metabolism of drugs, xenobiotics, and endogenous compounds in humans. Among the more than 50 CYP isoforms identified, five enzymes—CYP3A4, CYP2D6, CYP2C9, CYP1A2, and CYP2C19—account for over 90% of marketed drug metabolism and are the principal mediators of clinically significant drug–drug interactions (DDIs). Inhibition of these enzymes by co-administered drugs can precipitate elevated plasma concentrations of victim drugs, leading to exaggerated pharmacological effects or severe adverse reactions. Additionally, CYP enzymes play pivotal roles in the bioactivation of procarcinogens and the metabolism of chemotherapeutic agents, making CYP inhibition a strategic target in oncology research. Profacgen provides a comprehensive CYP inhibition screen assay using LC-MS/MS that delivers quantitative, reproducible inhibition data to support DDI risk assessment, lead optimization, and regulatory submission.
Figure 1. CYP inhibition assay. (Dinger et al., 2014)
Introduction to CYP Inhibition Studies
The clinical relevance of CYP-mediated drug metabolism has been firmly established through decades of pharmacokinetic research and regulatory guidance. The U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) mandate in vitro CYP inhibition studies as a prerequisite for investigational new drug (IND) applications, requiring sponsors to evaluate the inhibitory potential of new molecular entities (NMEs) against major CYP isoforms. These studies inform critical decisions regarding clinical DDI study design, dosing regimen adjustments, and contraindication labeling.
CYP inhibition is classified into two principal categories:
Reversible inhibition: Rapid, equilibrium-based inhibition resulting from direct binding of the inhibitor to the CYP active site. Subtypes include competitive, non-competitive, and uncompetitive inhibition, distinguished by their effects on apparent Km and Vmax.
Time-dependent inhibition (TDI): Progressive loss of enzymatic activity during pre-incubation with the inhibitor, typically arising from mechanism-based inactivation (MBI) or metabolite-intermediate complex (MIC) formation. TDI poses heightened DDI risk because the inhibitory effect persists even after the inhibitor is cleared from systemic circulation.
Profacgen employs a validated LC-MS/MS platform to detect changes in the concentration of FDA-recommended probe substrates and their specific metabolites, enabling precise quantification of CYP inhibition. Probe substrates used in our assays include midazolam (CYP3A4), dextromethorphan (CYP2D6), diclofenac (CYP2C9), phenacetin (CYP1A2), mephenytoin (CYP2C19), tolbutamide (CYP2C9), and warfarin (CYP2C9/1A2), as recommended in FDA guidance documents.
Figure 2. CYP inhibition assay using LC-MS/MS. (Kahma et al., 2021)
Major CYP Isoforms
Profacgen's CYP inhibition screen focuses on the five isoforms most frequently implicated in drug metabolism and DDI liability:
Highest abundance in liver and intestine; involved in metabolism of >50% of marketed drugs; strong DDI liability with inhibitors (e.g., ketoconazole, ritonavir)
Reversible inhibition assays evaluate the immediate, equilibrium-dependent reduction in CYP activity upon co-incubation of the test compound with enzyme and substrate. Profacgen performs single-point inhibition screens (typically at 10 µM) for rapid triage, followed by full IC50 determinations (10-point, duplicate or triplicate) for compounds exceeding a predefined inhibition threshold (e.g., >50% at 10 µM). The assay configuration distinguishes competitive, non-competitive, and mixed inhibition through Dixon plot analysis or substrate-dependent IC50 shifts.
Time-Dependent Inhibition (TDI)
TDI assays assess whether the test compound undergoes CYP-mediated bioactivation to a reactive intermediate that covalently modifies the apoprotein or prosthetic heme, resulting in irreversible or quasi-irreversible inactivation. The standard two-step TDI protocol involves:
Step 1 (Pre-incubation): Test compound + CYP enzyme + NADPH for 0, 15, or 30 minutes
Step 2 (Activity assessment): Dilution into probe substrate + fresh NADPH; residual activity measured by metabolite formation
A time-dependent reduction in residual activity (IC50 shift ≥ 1.5-fold) indicates TDI liability. For confirmed TDI, kinact (maximum inactivation rate constant) and KI (concentration for half-maximal inactivation) are determined through progress curve analysis, enabling prediction of clinical DDI magnitude via the [I]/KI decision framework.
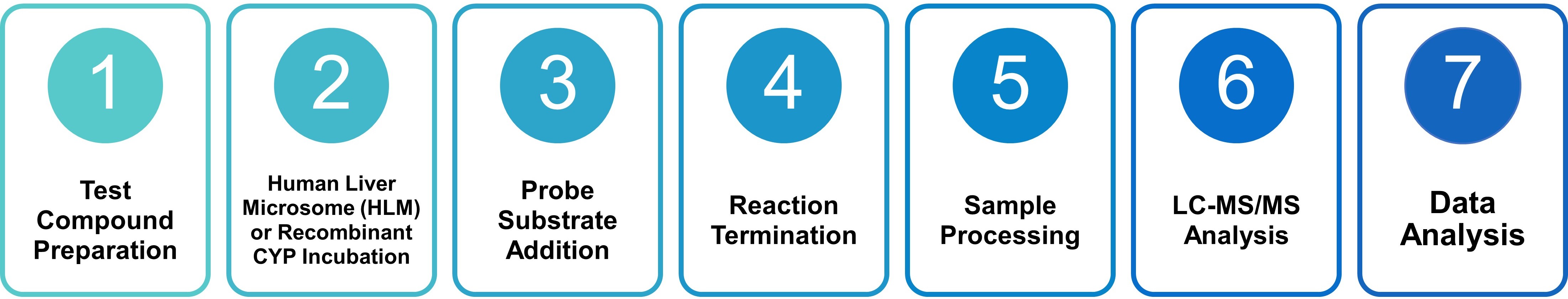
LC-MS/MS-Based Workflow
Liquid chromatography–tandem mass spectrometry (LC-MS/MS) is the analytical gold standard for CYP inhibition assays, offering unmatched sensitivity, selectivity, and dynamic range for the simultaneous quantification of probe substrates and their metabolites in complex biological matrices. Profacgen's workflow adheres to FDA and EMA guidance for in vitro DDI studies:
Test Compound Preparation: The test article (NME or candidate compound) is dissolved in an appropriate solvent (typically DMSO) and serially diluted across a concentration range (e.g., 0.01–100 µM) to generate a full dose–response relationship.
Human Liver Microsome (HLM) or Recombinant CYP Incubation: Pooled human liver microsomes (HLMs) or baculovirus-expressed recombinant CYP enzymes (with co-expressed NADPH-P450 reductase) are incubated with the test compound and NADPH-regenerating system at 37 °C. For TDI assessment, a pre-incubation step (typically 0–30 minutes) is included prior to substrate addition.
Probe Substrate Addition: Following pre-incubation (for TDI) or immediately (for reversible inhibition), FDA-recommended probe substrates are added at concentrations approximating their respective Km values to ensure maximal metabolic flux and sensitivity to inhibition.
Reaction Termination: At predetermined time points (typically 5–30 minutes), reactions are quenched with ice-cold organic solvent (e.g., acetonitrile or methanol) containing internal standards (stable isotope-labeled analogs) for accurate quantification.
Sample Processing: Precipitated proteins are removed by centrifugation, and supernatants are transferred to LC-MS/MS vials. Automated liquid handling ensures precision and traceability.
LC-MS/MS Analysis: Chromatographic separation is achieved on a C18 reverse-phase column with gradient elution. Detection is performed using triple-quadrupole mass spectrometers operating in multiple reaction monitoring (MRM) mode, with transitions optimized for each substrate–metabolite pair. Internal standard calibration curves ensure quantitative accuracy.
Data Analysis: Metabolite peak areas are normalized to internal standards and converted to concentrations using calibration curves. Percent inhibition is calculated relative to vehicle (DMSO) controls and fitted to a four-parameter logistic model to derive IC50 values. For TDI, the shift in IC50 following pre-incubation (IC50 shift) and the kinact/KI parameters are determined.
CYP inhibition data generated by LC-MS/MS are integral to multiple stages of pharmaceutical research and development:
Drug–drug interaction (DDI) risk assessment: Early identification of CYP inhibition liability guides clinical DDI study design, dosing recommendations, and labeling. Regulatory agencies require in vitro CYP inhibition data prior to first-in-human studies.
Lead optimization: Structure–activity relationship (SAR) campaigns leverage CYP IC50 data to engineer out metabolic liabilities while preserving target potency. Medicinal chemists use these data to prioritize analogs with minimal off-target CYP inhibition.
Regulatory submission support: Validated CYP inhibition studies conducted under Good Laboratory Practice (GLP) or GLP-like conditions provide the in vitro evidence required for IND, NDA, and MAA filings.
Personalized medicine and polypharmacy: Understanding the CYP inhibition profile of a drug candidate informs co-medication strategies, contraindication definitions, and patient population-specific dosing in populations with polymorphic CYP expression.
Oncology research: CYP-mediated bioactivation of procarcinogens and metabolism of chemotherapeutic agents (e.g., tamoxifen, cyclophosphamide) makes CYP inhibition relevant to both cancer prevention and therapeutic optimization.
Deliverables
Upon project completion, clients receive a comprehensive, audit-ready data package including:
IC50 values: Potency parameters for each CYP isoform with replicate statistics, 95% confidence intervals, and Hill slope coefficients
Inhibition curves: Publication-quality dose–response plots with four-parameter logistic fits and raw data overlays
Metabolite quantification: Absolute or relative metabolite concentrations (e.g., 1′-hydroxymidazolam, dextrorphan, 4′-hydroxydiclofenac) with internal standard calibration and quality control recoveries
TDI assessment: IC50 shift data, kinact/KI parameters (if applicable), and mechanistic classification (reversible vs. irreversible vs. quasi-irreversible)
Study report: Detailed protocol description, reagent qualifications, instrument calibration records, statistical analysis, and regulatory-compliant summary prepared by our DMPK scientists
Service Advantages
LC-MS/MS Analytical Excellence: Accurate, sensitive, and reproducible quantification compared to MEIA and other conventional methods; ability to identify and quantify trace compounds in complex sample matrices
Comprehensive Isoform Coverage: Standard panel includes CYP3A4, CYP2D6, CYP2C9, CYP1A2, and CYP2C19, with expansion to CYP2B6, CYP2E1, and CYP2A6 available
Regulatory Alignment: Assays designed in strict accordance with FDA (2020) and EMA (2012) DDI guidance documents; GLP-capable upon request
Dual Inhibition Assessment: Simultaneous evaluation of reversible and time-dependent inhibition to capture both immediate and latent DDI risks
Experienced DMPK Team: Deep expertise in in vitro drug metabolism, enzyme kinetics, and regulatory submission strategies
Integrated Service Portfolio: Seamless extension to CYP induction studies, reaction phenotyping, metabolite identification, and in vitro–in vivo extrapolation (IVIVE)
Representative Case Studies
Case 1: Reversible CYP3A4 Inhibition Assessment for an Oncology Candidate
Background:
A pharmaceutical sponsor required CYP inhibition profiling for a novel kinase inhibitor entering Phase I clinical trials. Preliminary data suggested potential CYP3A4 liability, necessitating a comprehensive in vitro assessment to inform clinical DDI study design and dosing recommendations.
Our Solution:
Profacgen performed a full reversible inhibition screen against all five major CYP isoforms using pooled HLMs and FDA-recommended probe substrates. For CYP3A4, both midazolam 1′-hydroxylation and testosterone 6β-hydroxylation were monitored to capture substrate-dependent inhibition differences. A 10-point IC50 curve was generated in duplicate with ketoconazole as the positive control.
Final Results:
The compound exhibited potent, competitive inhibition of CYP3A4 (IC50 = 0.8 µM) with minimal activity against CYP2D6, CYP2C9, CYP1A2, and CYP2C19 (IC50 > 50 µM). Dixon plot analysis confirmed competitive inhibition kinetics. These data supported the design of a clinical DDI study with a sensitive CYP3A4 substrate and informed the investigator brochure and regulatory submission.
Case 2: Time-Dependent Inhibition (TDI) Characterization of an Antifungal Agent
Background:
An antifungal drug candidate demonstrated unexpected clinical DDI liability in early Phase II studies, with elevated exposure to co-administered midazolam persisting beyond the compound's elimination half-life. The sponsor required mechanistic elucidation to determine whether TDI was responsible.
Our Solution:
Profacgen conducted a two-step TDI assay using recombinant CYP3A4 and HLMs. The test compound was pre-incubated with enzyme and NADPH for 0, 15, and 30 minutes, followed by dilution and assessment of residual midazolam 1′-hydroxylation activity. A control incubation without NADPH was included to rule out non-specific protein binding artifacts.
Final Results:
A 3.2-fold IC50 shift was observed following 30-minute pre-incubation with NADPH, confirming mechanism-based CYP3A4 inactivation. Progress curve analysis yielded kinact = 0.12 min−1 and KI = 4.5 µM. These parameters predicted a clinically relevant DDI ([I]/KI > 0.1), prompting the sponsor to reformulate the compound and implement a contraindication with strong CYP3A4 substrates.
Frequently Asked Questions (FAQs)
Q: What is the difference between reversible and time-dependent CYP inhibition?
A: Reversible inhibition is an equilibrium process in which the inhibitor binds to the CYP active site without permanent modification; activity is restored upon inhibitor dilution or removal. Time-dependent inhibition (TDI) involves CYP-mediated bioactivation to a reactive metabolite that covalently modifies the enzyme, resulting in irreversible or quasi-irreversible inactivation. TDI carries greater clinical DDI risk because the inhibitory effect persists after the parent drug is cleared.
Q: Why is LC-MS/MS preferred over other methods for CYP inhibition assays?
A: LC-MS/MS offers superior sensitivity, selectivity, and dynamic range compared to microplate enzyme immunoassay (MEIA), fluorescence, or radiometric methods. It enables simultaneous quantification of multiple probe substrates and metabolites in a single analytical run, reduces matrix interference, and provides definitive structural confirmation through mass transitions. These advantages translate to lower limits of quantification, improved reproducibility, and regulatory acceptance.
Q: What probe substrates does Profacgen use for CYP inhibition studies?
A: We employ FDA-recommended probe substrates including phenacetin (CYP1A2), diclofenac (CYP2C9), mephenytoin (CYP2C19), dextromethorphan (CYP2D6), and midazolam (CYP3A4). These substrates exhibit high CYP selectivity, well-characterized Km values, and clinically validated metabolic pathways, ensuring regulatory compliance and data interpretability.
Q: Can you predict clinical DDI magnitude from in vitro CYP inhibition data?
A: Yes. For reversible inhibitors, the [I]/IC50 or [I]/Ki ratio is used to estimate the fold-change in AUC of a sensitive substrate, with thresholds defined in FDA guidance (e.g., [I]/IC50 ≥ 0.1 suggests a clinical DDI). For TDI, the [I]/KI ratio combined with kinact is used in the mechanistic static model or dynamic PBPK modeling to predict DDI magnitude. Profacgen provides these extrapolations as part of our integrated DMPK service.
Q: How long does a standard CYP inhibition screen take?
A: A standard five-isoform single-point inhibition screen can be completed within 2–3 weeks. Full IC50 determinations with TDI assessment require 4–6 weeks, depending on compound number and replicate requirements. Expedited timelines are available for time-critical regulatory submissions.
Q: Do you offer CYP induction studies in addition to inhibition assays?
A: Yes. Profacgen provides integrated in vitro drug metabolism services including CYP induction (mRNA and enzyme activity endpoints in human hepatocytes), reaction phenotyping, metabolite identification, and in vitro–in vivo extrapolation (IVIVE). These services can be bundled with CYP inhibition studies for a comprehensive DDI risk assessment package.
References:
Dinger J, Meyer MR, Maurer HH. Development of an in vitro cytochrome P450 cocktail inhibition assay for assessing the inhibition risk of drugs of abuse. Toxicology Letters. 2014;230(1):28-35. doi:10.1016/j.toxlet.2014.08.004
Kahma H, Aurinsalo L, Neuvonen M, et al. An automated cocktail method for in vitro assessment of direct and time-dependent inhibition of nine major cytochrome P450 enzymes – application to establishing CYP2C8 inhibitor selectivity. European Journal of Pharmaceutical Sciences. 2021;162:105810. doi:10.1016/j.ejps.2021.105810
Online Inquiry
Fill out this form and one of our experts will respond to you within one business day.
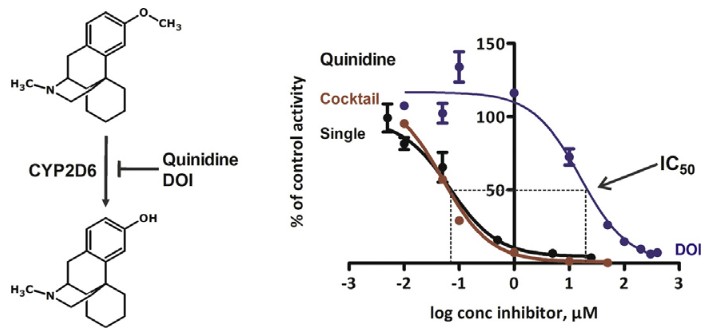 Figure 1. CYP inhibition assay. (Dinger et al., 2014)
Figure 1. CYP inhibition assay. (Dinger et al., 2014)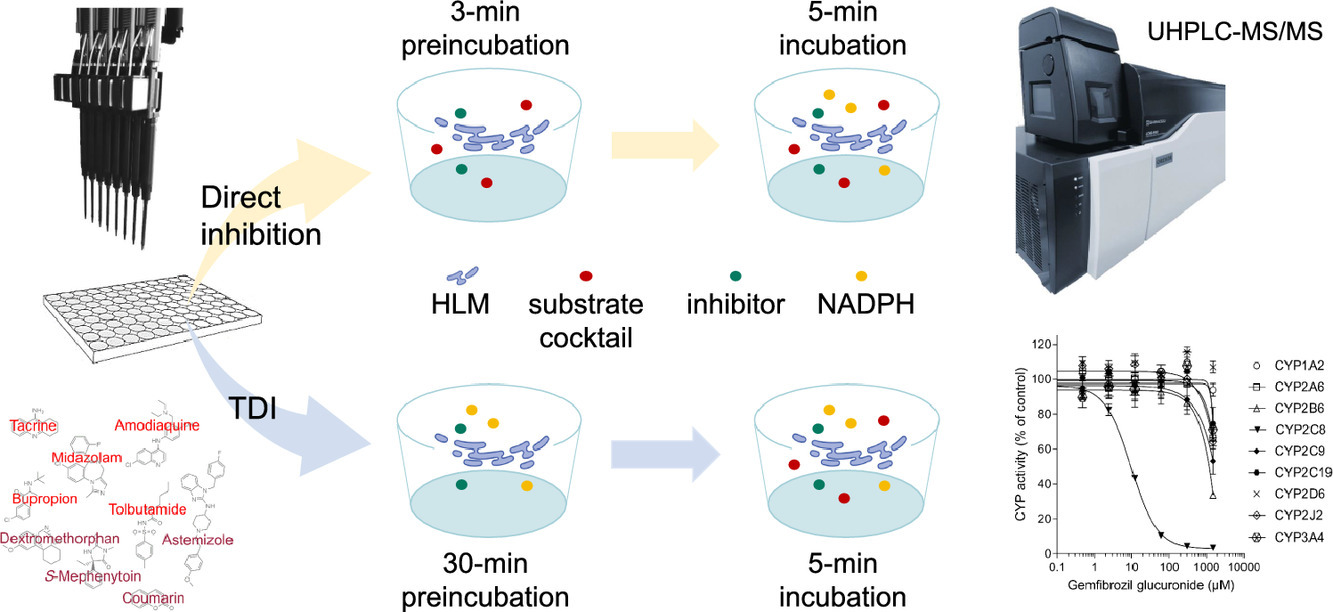 Figure 2. CYP inhibition assay using LC-MS/MS. (Kahma et al., 2021)
Figure 2. CYP inhibition assay using LC-MS/MS. (Kahma et al., 2021)