We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our
Privacy Policy
Receptor-ligand binding is the fundamental molecular event underlying signal transduction, immune recognition, and pharmacological action. Quantitative characterization of these interactions—determining affinity, selectivity, kinetics, and stoichiometry—is essential for drug discovery, therapeutic optimization, and mechanistic understanding of cellular communication. Profacgen provides comprehensive Receptor-Ligand Binding Assay services utilizing radioligand, fluorescence, and luminescence detection platforms to deliver precise binding parameters across diverse receptor classes, including G protein-coupled receptors (GPCRs), receptor tyrosine kinases, nuclear receptors, ion channels, and cytokine receptors. Our integrated approach combines classical pharmacological methodology with advanced detection technologies, supporting projects from early-stage target validation through regulatory-compliant therapeutic characterization.
Background: Principles of Receptor-Ligand Binding
Receptor-ligand interactions are governed by the law of mass action, where binding equilibrium is determined by the association rate constant (kon), dissociation rate constant (koff), and the resulting equilibrium dissociation constant (KD = koff/kon). This KD value—representing the ligand concentration at which 50% of receptors are occupied—serves as the primary quantitative descriptor of binding affinity and is inversely related to the equilibrium association constant (KA = 1/KD).
Figure 1. The kinetic and equilibrium state of a receptor and ligand binding event. (Wang and Fa, 2016)
Binding Models
Profacgen employs mathematically rigorous binding models tailored to the mechanistic complexity of each receptor-ligand system:
Model
Mathematical Basis
Information Content
Applications
Saturation Binding (Direct Binding)
Specific binding = Bmax × [L] / (KD + [L]); total binding minus nonspecific binding (determined in presence of excess unlabeled competitor)
KD (equilibrium dissociation constant), Bmax (receptor density or binding site concentration), Hill slope (cooperativity indicator)
Affinity determination, receptor expression quantification, comparative pharmacology across cell lines or tissues
Competition Binding (Displacement)
IC50 determination from sigmoidal displacement of a fixed tracer concentration by increasing unlabeled competitor; Ki calculated by Cheng-Prusoff correction: Ki = IC50 / (1 + [tracer]/KD,tracer)
Ki (inhibitory dissociation constant), binding site selectivity, allosteric modulation detection through shifts in tracer KD
kon (on-rate), koff (off-rate), residence time (τ = 1/koff), mechanism of association
Drug-target residence time optimization, mechanism-of-action differentiation, slow-offrate therapeutic design
Allosteric Modulation
Modulator effects on both tracer affinity (α factor) and efficacy (β factor) quantified through global fitting of tracer saturation curves at multiple modulator concentrations
GPCR allosteric drug discovery, bitopic ligand characterization, probe-dependent pharmacology assessment
Non-specific binding—defined as radioligand or tracer binding that cannot be displaced by pharmacologically relevant concentrations of unlabeled competitor—must be rigorously quantified and subtracted from total binding to yield specific binding parameters. Profacgen employs multiple negative control strategies, including excess unlabeled ligand, structurally unrelated competitors, and receptor knockout cell lines, to ensure accurate specific binding determination.
What We Offer: Assay Services
Profacgen provides a comprehensive portfolio of receptor-ligand binding assays configurable to diverse receptor families, ligand chemistries, and project objectives.
Detection Platforms
Radioligand Binding
Gold-standard detection using 3H, 125I, or 14C-labeled ligands with scintillation proximity assay (SPA), filtration, or homogeneous scintillation counting. Unmatched sensitivity for low-abundance receptors and direct KD determination without fluorescent probe modification artifacts.
Fluorescence Detection
Fluorescently labeled ligand binding detected by fluorescence polarization (FP), fluorescence intensity (FI), time-resolved Förster resonance energy transfer (TR-FRET), and fluorescence correlation spectroscopy (FCS). Compatible with high-throughput screening and live-cell imaging formats.
Luminescence Detection
Nanoluciferase-tagged ligands and bioluminescence resonance energy transfer (BRET)-based proximity assays for ultra-sensitive binding detection in living cells. Low background and high dynamic range enable single-molecule sensitivity and real-time kinetic monitoring.
Assay Formats by Receptor Class
Our platform accommodates the structural and mechanistic diversity of major therapeutic receptor families:
G Protein-Coupled Receptors (GPCRs): Membrane preparation-based radioligand binding, whole-cell fluorescence assays, and bioluminescence resonance energy transfer (BRET)-based live-cell binding assays for class A, B, C, and frizzled receptors; orthosteric and allosteric site mapping using probe-dependent competition panels
Receptor Tyrosine Kinases (RTKs): 125I-growth factor displacement, fluorescent peptide binding to extracellular domains, and TR-FRET-based dimerization assays; receptor internalization kinetics by radioligand acid wash protocols
Nuclear Receptors: Fluorescent ligand binding to purified ligand-binding domains (LBDs), scintillation proximity assays with 3H-steroid hormones, and fluorescence polarization for coactivator peptide recruitment
Ion Channels: Radioligand binding to voltage-gated and ligand-gated channel allosteric sites; fluorescent neurotransmitter analog binding for competitive displacement; state-dependent binding using electrophysiology-coupled protocols
Cytokine and Chemokine Receptors: 125I-cytokine saturation and competition, biotinylated ligand-avidin capture formats, and BRET-based live-cell receptor occupancy assays for biologic therapeutic development
Specialized Binding Services
Thermodynamic Binding Analysis: Isothermal titration calorimetry (ITC) and differential scanning fluorimetry for enthalpy-driven optimization and binding mechanism deconvolution
Cellular Target Engagement: Energy transfer–based live-cell binding assays and CETSA (cellular thermal shift assay) formats confirming intracellular target occupancy by membrane-permeable compounds
Binding Kinetics by Surface Plasmon Resonance (SPR): Label-free real-time association and dissociation monitoring for purified receptor extracellular domains and soluble ligands
Receptor Occupancy Assays: Ex vivo radioligand binding to tissue homogenates for in vivo pharmacodynamic biomarker development
Service Procedure
Profacgen executes receptor-ligand binding projects through a standardized, quality-controlled workflow ensuring data integrity, reproducibility, and regulatory compliance.
Multi-Platform Detection Flexibility: Radioligand, fluorescence, luminescence, and label-free SPR capabilities under one roof enable platform selection optimized for each receptor's biochemical properties and each ligand's detection compatibility
Comprehensive Receptor Coverage: Established protocols for GPCRs, RTKs, nuclear receptors, ion channels, cytokine receptors, and emerging target classes with validated reference ligand databases
Advanced Pharmacological Modeling: Beyond IC50 reporting, we provide Ki determination, kinetic rate constant extraction, allosteric parameter estimation, and thermodynamic decomposition to guide mechanism-based drug design
Regulatory-Compliant Operations: GLP-aligned assay validation (ICH Q2(R1)), instrument qualification, analyst training documentation, and full audit trails supporting IND and NDA submissions
High-Throughput Screening Execution: Automated liquid handling, acoustic dispensing, and integrated data pipelines for single-concentration primary screens and multi-point dose-response confirmation campaigns
Representative Case Studies
Case 1: Allosteric Site Mapping for a Class B GPCR Glucagon-like Peptide-1 Receptor (GLP-1R)
Background:
A pharmaceutical company developing small molecule allosteric modulators of GLP-1R for type 2 diabetes required mechanistic understanding of compound binding site location relative to the orthosteric peptide binding pocket. Standard competition binding using radiolabeled GLP-1 showed complex, non-competitive displacement patterns suggesting allosteric interaction, but could not distinguish between negative allosteric modulators (NAMs) and neutral allosteric ligands (NALs).
Our Solution:
Profacgen implemented a comprehensive allosteric pharmacology platform: (1) 125I-GLP-1 saturation binding at multiple allosteric compound concentrations to quantify affinity modulation (α factor); (2) 125I-exendin-4 (orthogonal probe) competition to assess probe dependence; (3) TR-FRET-based coactivator peptide recruitment to measure efficacy modulation (β factor); and (4) kinetic dissociation assays to detect allosteric effects on tracer off-rate. Global fitting of the multi-probe dataset was performed using the allosteric ternary complex model.
Final Results:
The analysis revealed two distinct allosteric chemotypes: one acting as a NAM (α = 0.3, β = 0.1) that reduced both GLP-1 affinity and efficacy, and another as a pure NAL (α = 1.0, β = 0.8) that enhanced coactivator recruitment without affecting peptide binding. The NAL chemotype advanced to lead optimization as a potential GLP-1R biased agonist with reduced receptor desensitization, while the NAM series was deprioritized based on unfavorable mechanism-of-action profiling.
Case 2: Kinetic Binding Optimization for a CNS-Targeted D2 Dopamine Receptor Antagonist
Background:
A neuroscience drug discovery program sought a D2 receptor antagonist with rapid onset for acute psychosis management but reduced motor side effect liability through transient receptor occupancy. Biochemical Ki values from standard equilibrium binding failed to predict in vivo duration of action, as compounds with identical affinity exhibited markedly different clinical profiles in early pharmacokinetic/pharmacodynamic (PK/PD) studies.
Our Solution:
Profacgen conducted kinetic binding characterization of eight lead compounds using radioligand association-dissociation protocols on D2L receptor membranes. Association kinetics were measured by 3H-raclopride binding time courses at multiple concentrations, while dissociation was initiated by infinite dilution or excess unlabeled competitor addition. Residence time (τ = 1/koff) was calculated for each compound and correlated with rat microdialysis extracellular dopamine levels and catalepsy scores.
Final Results:
A strong inverse correlation emerged between D2 receptor residence time and catalepsy incidence (R² = 0.87), while antipsychotic efficacy in amphetamine-induced locomotion models correlated with peak receptor occupancy rather than residence time. Compounds with τ < 10 minutes provided sufficient acute antagonism with rapid offset, minimizing D2 receptor upregulation and motor side effects. The kinetic binding data redirected lead optimization toward fast-offrate candidates, accelerating identification of a compound that entered Phase I with a differentiated clinical profile.
Q: What is the difference between KD, Ki, and IC50 in binding assays?
A: KD is the equilibrium dissociation constant determined directly from saturation binding experiments, representing the concentration of ligand required to occupy 50% of receptors at equilibrium. It is a true thermodynamic constant independent of assay conditions. IC50 is the concentration of unlabeled competitor that displaces 50% of a fixed tracer concentration in a competition binding assay; it depends on tracer concentration and affinity. Ki is the calculated inhibitory dissociation constant derived from IC50 using the Cheng-Prusoff equation (Ki = IC50 / (1 + [tracer]/KD,tracer)), correcting for tracer occupancy to yield a tracer-independent affinity estimate comparable to KD. For accurate pharmacological comparison across studies, KD or Ki should be reported rather than raw IC50 values.
Q: When should radioligand binding be used instead of fluorescent or luminescent methods?
A: Radioligand binding remains the gold standard when: (1) receptor expression is low and maximum sensitivity is required; (2) the ligand lacks suitable functional groups for fluorophore conjugation without affinity loss; (3) direct KD determination is needed without potential artifacts from probe modification; (4) regulatory agencies specifically require radioligand data for registration dossiers; or (5) studying lipophilic ligands where fluorescent analogs exhibit high nonspecific binding. Fluorescence and luminescence methods are preferred for high-throughput screening, live-cell imaging, real-time kinetics, and when radioisotope handling restrictions apply. Profacgen selects the optimal platform based on target properties, ligand chemistry, and project objectives.
Q: How do you distinguish specific from nonspecific binding in receptor assays?
A: Specific binding is defined as the component of total tracer binding that can be displaced by excess unlabeled competitor with pharmacological relevance to the receptor under study. Nonspecific binding represents tracer association with non-receptor sites (lipid membranes, plastic surfaces, filter matrices) that is not competitively inhibitable. Profacgen quantifies nonspecific binding using: (1) 100–1000× excess unlabeled specific ligand; (2) structurally unrelated competitors at high concentration; (3) receptor knockout cell lines or membranes; and (4) saturation binding analysis where nonspecific binding is modeled as a linear function of free ligand concentration. Specific binding is calculated as total binding minus nonspecific binding, and should constitute >70% of total binding at KD for assay validity.
Q: Can binding assays detect allosteric modulation, or only orthosteric competition?
A: Binding assays are powerful tools for allosteric modulation detection when designed appropriately. Allosteric modulators alter the affinity (α factor) and/or efficacy (β factor) of orthosteric ligands without competing for the same binding site. Key experimental signatures include: (1) incomplete displacement of tracer even at saturating modulator concentrations; (2) changes in tracer KD (saturation curve shifts) in the presence of modulator; (3) probe dependence, where modulator effects vary with different orthosteric tracers; and (4) altered tracer dissociation kinetics. Profacgen employs the allosteric ternary complex model for global analysis of multi-probe, multi-concentration datasets to quantify cooperativity factors and distinguish allosteric antagonism from neutral allosteric ligands and positive allosteric modulators (PAMs).
Q: What is the relationship between binding kinetics (kon, koff) and therapeutic efficacy?
A: While equilibrium affinity (KD = koff/kon) determines receptor occupancy at steady state, binding kinetics critically influence drug action in vivo where concentrations fluctuate due to absorption, distribution, metabolism, and excretion. The dissociation rate constant koff determines receptor residence time (τ = 1/koff), which correlates with duration of pharmacological effect beyond plasma drug clearance. Slow-offrate compounds can maintain target engagement at sub-saturating plasma concentrations, potentially improving efficacy and reducing dosing frequency. Conversely, fast-offrate ligands may minimize side effects from excessive receptor blockade. Profacgen's kinetic binding services enable residence time optimization as a distinct drug design parameter independent of equilibrium affinity.
Q: What is the typical project timeline for receptor-ligand binding assay development?
A: Standard assay development timelines are: (1) 3–4 weeks for established receptor-ligand pairs with validated tracers, including membrane preparation, tracer KD confirmation, and competition assay optimization; (2) 6–8 weeks for novel receptor targets requiring expression system evaluation, tracer identification, and assay format selection; (3) 8–10 weeks for allosteric modulation or kinetic binding characterization with multi-parameter global fitting; and (4) 10–12 weeks for full GLP-compliant assay validation with documented precision, accuracy, specificity, and robustness. High-throughput screening campaigns are executed on validated assays with 2–3 week turnaround for single-concentration primary screens and 4–6 weeks for multi-point dose-response confirmation.
References:
Montet X, Yuan H, Weissleder R, Josephson L. Enzyme-based visualization of receptor–ligand binding in tissues. Laboratory Investigation. 2006;86(5):517-525. doi:10.1038/labinvest.3700404
Wang DS, Fan SK. Microfluidic surface plasmon resonance sensors: from principles to point-of-care applications. Sensors. 2016;16(8):1175. doi:10.3390/s16081175
Online Inquiry
Fill out this form and one of our experts will respond to you within one business day.
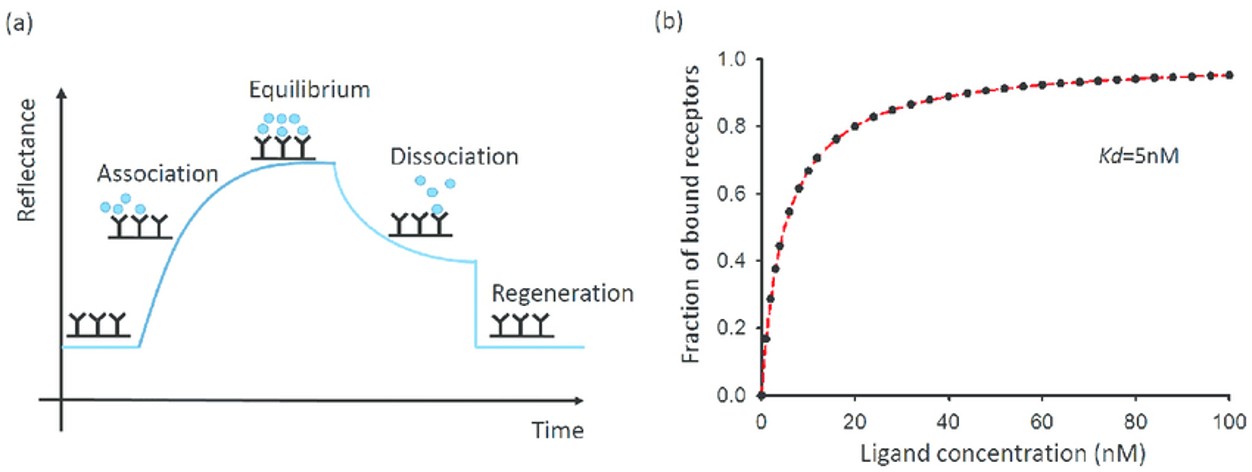 Figure 1. The kinetic and equilibrium state of a receptor and ligand binding event. (Wang and Fa, 2016)
Figure 1. The kinetic and equilibrium state of a receptor and ligand binding event. (Wang and Fa, 2016)
